Chapter: Basic Concept of Biotechnology : Macromolecules and Analytical Techniques
Electrophoresis - Analysis of biomolecules
Electrophoresis
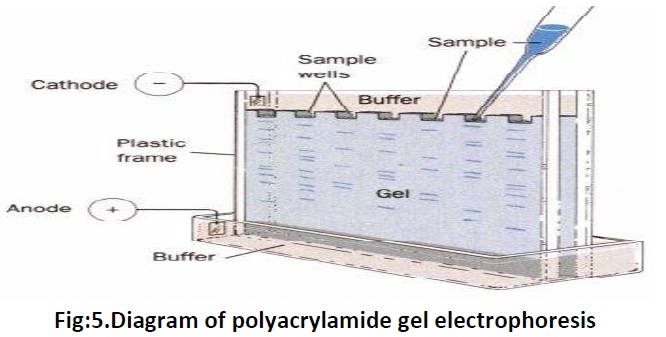
The molecules are separated on the basis of their charge and mass ratio in an electric field or in another words migration of charged ions in an electric field, this is the principle of electrophoresis. Several types of medium are present to perform the electrophoresis procedure via; Filter paper which is moistened with buffer and placed between electrodes and by which small molecules, amino acids and nucleotides can be separated. Polyacrylamide gelwhich are covalently cross linked and are casted in tubes or as slabs, they are used to separate proteins and nucleic acids. Agarose gel a highly purified polysaccharide derived from agar (extracted from seeweed), long sugar polymers held together by hydrogen and hydrophobic bonds but with no crosslinks are used to separate very large proteins, nucleic acids and nucleoproteins. Two types of gel electrophoresis include; one dimension and two dimension. One dimension includes SDS-PAGE, Native PAGE and Iso Electric Focussing (IEF). SDS-PAGE, the most widely used electrophoresis technique, separates proteins primarily by mass. Non denaturing PAGE, also called native PAGE, separates proteins according to their mass/charge ratio. The two-dimensional PAGE (2D-PAGE) separates proteins by isoelectric point in the first dimension and by mass in the second dimension.
Sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE).
SDS-PAGE is a method of gel electrophoresis to separate proteins based on the mass. Sodium dodecyl sulphate (SDS) is an anionic detergent that breaks up the interactions between proteins. This also disrupts secondary and tertiary protein structures by breaking hydrogen bonds and unfolding protein. SDS binds to protein in a ratio of one SDS molecule/two amino acids. SDS will give a total negative charge to all the protein molecules irrespective of their charges. The negatively charged proteins denatured by SDS are applied to one end of a layer of polyacrylamide gel submerged in a buffer. The buffer provides uniform pH and ions for conducting electric potential. Some of the common buffers used are Barbital buffer & Tris-EDTA, Tris-acetate-EDTA and Tris-borate-EDTA. When an electric current is applied across the gel, the negatively charged proteins migrate across the gel to the positive pole. The gel used for SDS-PAGE is made out of acrylamide which form cross-linked polymers of polyacrylamide. Standard gels are typically composed oftwo layers, one top most layer called the stacking gel and a lower layer called separating or resolving gel. The stacking layer contains a low percentage of acylamide and has low pH, while the acrylamide concentration of the separating gel varies according to the samples to be run and has higher pH. The difference in pH and acrylamide concentration at the stacking and separating gel provides better resolution and sharper bands in the separating gel. Short proteins will more easily fit through the pores in the gel and move fast, while larger ones will have more difficulty. Due to differential migration based on their size, smaller proteins move farther down the gel, while larger ones stay closer to the point of origin. After a given period of time, proteins might have separated roughly according to their sizes. Standard proteins were also run along the side of the test sample for molecular weight determination. Following electrophoresis, the gel may be stained with Coomassie Brilliant Blue or silver stain to visualize the separated proteins. After staining, different proteins will appear as distinct bands within the gel according to their sizes (and therefore by molecular weights).The molecular weight of a protein in the band can be estimated by comparing it with the marker proteins of known molecular weights. The relative mobility of each protein band is calculated as,

Native PAGE is used to separate proteins in their native states according to difference in their charge density. No denaturants will be added to the gel and buffer. It is used for preparation of purified and active proteins. In native PAGE the mobility depends on both the protein's charge and its hydrodynamic size. The charge depends on the amino acid composition of the protein as well as post- translational modifications. Iso electric Focusing (IEF) is a technique for separating different molecules bytheir electric charge differences. In this technique proteins are separated by electrophoresis in a pH gradient based on their isoelectric point (pl). A pH gradient is generated in the gel and an electric potential is applied across the gel. At all pHs other than their isoelectric point, proteins will be charged. If they are positively charged, they will move towards the more negative end of the gel and if they are negatively charged they will move towards the more positive end of the gel. At its isoelectric point, since the protein molecule carries no net charge it focuses into a sharp band. In two dimensional electrophoresis the first step is to perform IEF and then SDS-PAGE. Proteins having same molecular weight or same pI are also resolved. This technique can be used for analysis of complex mixture of proteins, partial characterization of proteins. Use of the electrophoresis include, to determine molecular weight of a peptide/protein, to identify protein, to determine sample purity, to identify existence of disulfide bonds, to quantify amounts of protein
One of the best uses of electrophoresis is its use in DNA and protein fingerprinting viz; Southern, Northern and Western blotting. Western blot (also called as immunoblot) is a technique to detect specifically one protein in a mixture of large number of proteins and to obtain information about the size and relative amounts of the protein present in different samples. At first, proteins are separated using SDS-polyacrylamide gel electrophoresis. Then they are movedonto a nitrocellulose membrane. The proteins retain the same pattern of separation they had on the gel.An antibody is then added to the solution which is able to bind to its specific protein and forms an antibody-protein complex with the protein of interest. Finally the nitrocellulose membrane is incubated with a secondary antibody, which is an antibody-enzyme conjugate that is directed against the primary antibody. The location of the antibody is revealed by incubating it with a substrate that the attached enzyme converts to a product that can be seen and followed and then photographed. In southern blotting genes can be localized by means of hybridization to radioactive DNA or RNA molecule (probe) which has a complementary sequence. Southern blotting was developed by Edwin Southern in 1975 it is also called as DNA blotting or hybridization. It is especially used to analyze the DNA or sometimes also called as DNA fingerprinting. In its first step the DNA from the source is extracted and cut into many pieces by using restriction enzymes. The segmented DNA are run in an agarose gel electrophoresis and stained with ehidium bromide. The gel is taken out and placed on neutralized nitrocellulose paper or nylon filter. Then to this 32p radio labeled complimentary DNA or RNA in a minimum solution volume at 68 oc of higher ionic strength is added which hybridizes with DNA, this is called hybridization. Then the paper is covered with X ray film or photographic plate which, when developed, reveals the position of the radioactive probe. The difference between northern and southern blotting is, in case of northern blotting which is used for the study of RNA, alkali is not used since the alkali hydrolyses the RNA instead formaldehyde is used. Since RNA doesn’t bind to nitrocellulose paper unless it is denatured so DBM or diazobenzyloxymethyl-paper is used. Northern blotting was developed by James Alwine, David Kemp and George Stark in 1977. With this technique quality and quantity of gene can be measured.
Related Topics