Chapter: Pharmaceutical Biotechnology: Fundamentals and Applications : Production and Downstream Processing of Biotech Compounds
Downstream Processing of Biotech Compounds
DOWNSTREAM
PROCESSING
Recovering a biological reagent from a cell culture supernatant is one
of the critical parts of the manufacturing procedure for biotech products and
purification costs typically outweigh those of the upstream part of the
production process. For the production of monoclonal antibodies, Protein A
resin accounts for some 10% of the cost while virus filtration can account for
some 40% of the cost (Gottschalk, 2006).
Usually, the product is available in a very dilute form, e.g., 10 to 200
mg/L, but concentrations up to 500 to 800 mg/L can be reached (Berthold and
Walter, 1994; Garnick et al., 1988). The prediction is that future development
in cell culture technology through application of genetics and proteomics will
result in product titers in the 5 to 10 g/L range which will challenge the
capacity of the downstream processing unit operations (Werner, 2005).
A concentration step is often required to reduce handling volumes for
further purification. Usually, the product subsequently undergoes a series of
purification steps. The first step traditionally captures and initially
purifies the product, the subsequent steps remove the bulk of the contaminants,
and a final step removes all trace contaminants and variant forms of the
molecule. Alternatively, the reverse strategy, where the main contaminants are
captured and the product is purified in subsequent steps, might result in a
more economic process, especially if the product is not excreted from the
cells. In the former case the product will not represent more than 1% to 5% of
total cellular protein and aspecific binding of the bulk of the protein in a
product specific capture step will ruin its efficiency. If the bulk
contaminants can be removed first, the specific capture step will be more
efficient and smaller in size, thus cheaper, and chromato-graphic columns could
be used. After purification, additional steps like formulation and
sterilization are performed on the bulk product in order to obtain the required
stable final product. Formulation aspects will be dealt.
When designing a purification protocol, the possibility for scaling up
should be considered care-fully. A process that has been designed for small
quantities is most often not suitable for large quantities for both technical
and economic reasons. Developing a downstream process (i.e., the isolation and
purification of the desired product) to recover a biological protein in large
quantities occurs in two stages: design and scale-up.
Separating the impurities from the product protein requires a series of
purification steps (process design), each removing some of the impurities and
bringing the product closer to its final specification. In general, the
starting feedstock contains cell debris and/or whole-cell particulate material
that must be removed. Defining the major contaminants in the starting material
is helpful in the downstream process design. This includes detailed information
on the source of the material (e.g., bacterial or mammalian cell culture) and
major contaminants (e.g., albumin orproduct analogs). Moreover, the physical
character-istics of the product versus the known contaminants (thermal stability,
isoelectric point, molecular weight, hydrophobicity, density, specific binding
properties) largely determine the process design. Processes used for production
of therapeutics should be reproducible and reliable. Methods used for recovery
may expose the protein molecules to high physical stress (e.g., high
temperatures and extreme pH) which alter the protein properties leading to
appreciable loss in protein activity. Any substance that is used by injection
must be sterile and free from pyrogens below a certain level depending on the
product (limits are stated in the individual monographs which are to be
consulted, such as European Pharmacopoeia: less than 0.2 mg/kg/body mass for
intrathecal applica-tion). This necessitates aseptic techniques and proce-dures
throughout with clean air and microbial control of all materials and equipment.
During validation of the purification process it must also be demonstrated that
potential viral contaminants can be removed (Walter et al., 1992). The
purification matrices should be at least sanitizable or, if possible
steam-sterilizable. For depyrogenation, the purification material must
withstand either extended dry heat at 180 C or treatment with 1 to 2 M sodium
hydroxide. If any material in contact with the product inadvertently releases
compounds, these leachables must be analyzed and their removal by subsequent
purification steps must be demon-strated during process validation. The
increased use of plastic film based disposable production technol-ogy (e.g.,
sterile bags to store liquids and filter housings) has made these aspects more
significant in the last 5 years and suppliers have reacted by providing a
significant body of information regarding leachables and biocompatability for
typical solutions used during processing. This problem of leachables is
especially hampering the use of affinity chromato-graphy (see below) in the
production of pharmaceu-ticals for human use. On laboratory scale affinity
chromatography is an important tool for purification and the resulting product
might be used for toxicity studies, but for human use the removal of any
leached ligands has to be demonstrated. Because free affinity ligands will bind
to the product, the removal might be very cumbersome.
Scale-up is the term used to describe a number of processes employed in
converting a laboratory pro-cedure into an economical, industrial process.
During the scale-up phase, the process moves from the laboratory scale to the
pilot plant and finally to the production plant. The objective of scale-up is
to produce a product of high quality at a competitive price. Since costs of
downstream processing can be as high as 50% to 80% of the total cost of a
product,practical and economical ways of purifying the product should be used.
Superior protein purification methods hold the key to a strong market position
(Wheelwright, 1993).
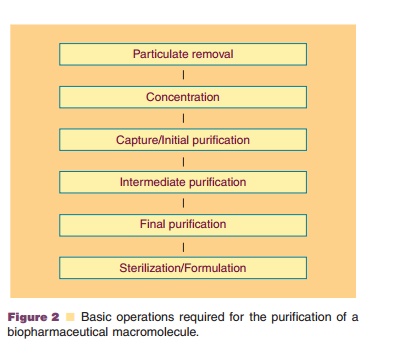
Basic operations required for a downstream purification process used for
macromolecules from biological sources are shown in Figure 2.
As mentioned before, the design of downstream processing is highly
product dependent. Therefore, each product requires a specific multistage
purifica-tion procedure (Sadana, 1989). The basic scheme as represented in
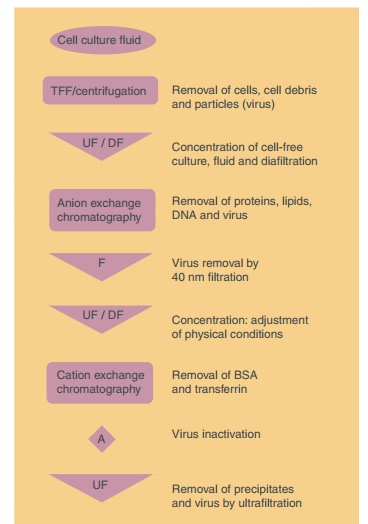
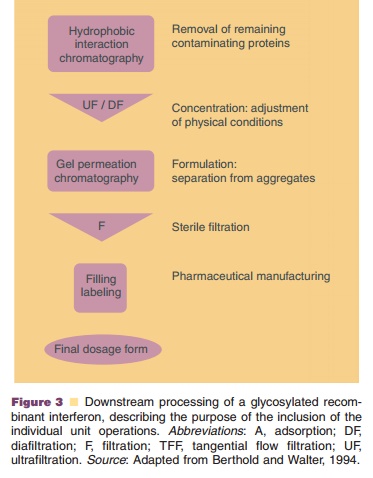
Figure 2 becomes complex. A typical example of a process flow for the downstream
processing is shown in Figure 3. This scheme represents the processing of a
glycosylated recombi-nant interferon (about 28 kDa) produced in mamma-lian
cells. The aims of the individual unit operations are described.
Once the volume and concentration of the product can be managed, the
main purification phase can start. A number of purification methods are
available to separate proteins on the basis of a wide variety of different
physicochemical criteria such as size, charge, hydrophobicity and solubility.
Detailed information about some separation and purification methods commonly
used in purification schemes is provided below.
Related Topics