Chapter: Pharmaceutical Biotechnology: Fundamentals and Applications : Production and Downstream Processing of Biotech Compounds
Chromatography - Downstream Processing of Biotech Compounds
Chromatography
In preparative chromatography systems molecular species are primarily separated based on differences in distribution between two phases, one which is the stationary phase (mostly a solid phase) and the other which moves. This mobile phase may be liquid or gaseous. Nowadays, almost all sta-tionary phases (fine particles providing a large surface area) are packed into a column. The mobile phase is passed through by pumps. Downstream purification protocols usually have at least two to three chromato-graphy steps. Chromatographic methods used in purification procedures of biotech products are listedin Table 5 and are briefly discussed in the following sections
Chromatographic Stationary Phases
Chromatographic procedures often represent the rate-limiting step in the overall downstream processing. An important primary factor governing the rate of operation is the mass transport into the pores of conventional packing materials. Adsorbents em-ployed include inorganic materials such as silica gels, glass beads, hydroxyapatite, various metal oxides (alumina) and organic polymers (cross-linked dex-trans, cellulose, agarose). Separation occurs by differ-ential interaction of sample components with the chromatographic medium. Ionic groups such as amines and carboxylic acids, dipolar groups such as carbonyl functional groups, and hydrogen bond-donating and accepting groups control the interaction of the sample components with the stationary phase and these functional groups slow down the elution rate if interaction occurs.
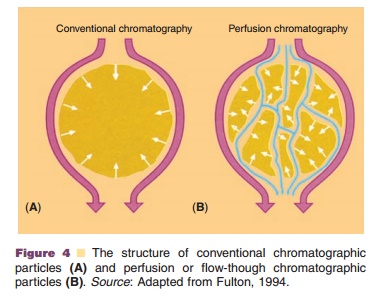
Chromatographic stationary phases for use on a large scale have improved considerably over the last decades. Hjerten et al. (1993) reported on the use of compressed acrylamide-based polymer structures. These materials allow relatively fast separations with good chromatographic performance. Another approach to the problems associated with mass transport in conventional systems is to use chromato-graphic particles that contain some large “through pores” in addition to conventional pores (Fig. 4). These flow-through or “perfusion chromatography” media enable faster convective mass transport into particles and allow operation at much higher speeds without loss in resolution or binding capacity (Afeyan et al., 1989; Fulton, 1994). Another development is the design of spirally wrapped columns containing the adsorption medium. This configuration permits high throughput, high capacity and good capture efficiency (Cartwright, 1987).
The ideal stationary phase for protein separation should possess a number of characteristics, among which are high mechanical strength, high porosity, no non-specific interaction between protein and the support phase, high capacity, biocompatibility and high stability of the matrix in a variety of solvents. The latter is especially true for columns used for the production of clinical materials that need to be cleaned, depyrogenized, disinfected and sterilized at regular intervals. High-performance liquid chromato-graphy (HPLC) systems fulfill many of these criteria. Liquid phases should be carefully chosen to minimize loss of biological activity resulting from the use of some organic solvents. In HPLC small pore size stationary phases that are incompressible are used. These particles are small, rigid and regularly sized (to provide a high surface area). The mobile liquid phase is forced under high pressure through the column material. Reversed-phase HPLC systems, using less polar stationary phases than the mobile phases can be effectively integrated into large-scale purification schemes of proteins and can serve both as a means of concentration and purification (Benedek and Swadesh, 1991).
In production environments columns which operate at relatively low back pressure are often used. They have the advantage that they can be used in equipment constructed from plastics which, unlike conventional stainless steel equipment, resists all buffers likely to be employed in the separation of biomolecules (consider the effect of leachables men-tioned earlier). These columns are commercially available and permit the efficient separation of proteins in a single run, making this an attractive unit operation in a manufacturing process. Results can be obtained rapidly and with high resolution. A new development is the use of stainless steel equipment that resists almost all chemicals used in protein purification including disinfection and ster-ilization media.
Unfortunately, HPLC equipment costs are high and this technology finds only limited application in large-scale purification schemes (Strickler and Gemski, 1987; Jungbauer and Wenisch, 1989).
Adsorption Chromatography
In adsorption chromatography (also called “normal phase” chromatography) the stationary phase is more polar than the mobile phase. The protein of interest selectively binds to a static matrix under one condi-tion and is released under a different condition (Chase, 1988). Adsorption chromatography methods enable high ratios of product load to stationary phase volume. Therefore, this principle is economically scalable.
Ion-Exchange Chromatography
Ion-exchange chromatography can be a powerful step at the beginning of a purification scheme. It can be easily scaled up. Ion-exchange chromatography can be used in a negative mode, i.e., the product flows through the column under conditions that favor the adsorption of contaminants to the matrix, while the protein of interest does not bind (Tennikova and Svec, 1993). The type of the column needed is determined by the properties of the proteins to be purified (e.g., isoelectric point and charge density). Anion exchan-gers bind negatively charged molecules and cation exchangers bind positively charged molecules. In salt-gradient ion-exchange chromatography, the salt con-centration in the perfusing elution buffer is increased continuously or in steps. The stronger the binding of an individual protein to the ion exchanger, the later it will appear in the elution buffer. Likewise, in pH-gradient chromatography, the pH is changed con-tinuously or in steps. Here, the protein binds at one pH and is released at a different pH. As a result of the heterogeneity in glycosylation, glycosylated proteins may elute in a relatively broad pH range (up to 2 pH units).
In order to simplify purification, a specific amino acid tail can be added to the protein at the gene level to create a “purification handle”. For example, a short tail consisting of arginine residues allows a protein to bind to a cation exchanger under conditions where almost no other cell proteins bind. However, this technique is only useful for laboratory scale isolation of the product and cannot be used for production scale due to regulatory problems related to the removal of the arginine or other specific tag from the protein.
(Immuno)Affinity Chromatography
Affinity chromatography is based on highly specific interactions between an immobilized ligand and the protein of interest. Affinity chromatography is a very powerful method for the purification of proteins. Under physiological conditions the protein binds to the ligand. Extensive washing of this matrix will remove contaminants and the purified protein can be recovered by the addition of ligands competing for the stationary phase binding sites or by changes in physical conditions (such as low or high pH of the eluent) which greatly reduce the affinity. Examples of affinity chromatography include the purification of glycoproteins, which bind to immobilized lectins and the purification of serine proteases with lysine binding sites, which bind to immobilized lysine. In these cases a soluble ligand (sugar or lysine, respec-tively) can be used to elute the required product under relatively mild conditions. Another example is the use of the affinity of protein A and protein G for antibodies. Protein A and protein G have a high affinity for the Fc portions of many immunoglobulins from various animals. Protein A and G matrices are commercially obtained with a high degree of purity. For the purification of e.g., hormones or growth factors, the receptors or short peptide sequence that mimic the binding site of the receptor molecule can be used as affinity ligands. Some proteins show highly selective affinity for certain dyes commercially avail-able as immobilized ligands on purification matrices. When considering the selection of these ligands for pharmaceutical production, one must realize that some of these dyes are carcinogenic and that a fraction may leach out during the process.
An interesting approach to optimize purification is the use of a gene that codes not only for the desired protein, but also for an additional sequence that facilitates recovery by affinity chromatography. At a later stage the additional sequence is removed by a specific cleavage reaction. As mentioned before, this is a complex process that needs additional purification steps.
In general, use of affinity chromatography in the production process for therapeutics leads tocomplications during validation of the removal of free ligands or protein extensions. Consequently, this technology is rarely used in the industry.
The specific binding of antibodies to their epitopes is used in immunoaffinity chromatography (Chase, 1993; Kamihira et al., 1993). This technique can be applied for purification of either the antigen or the antibody. The antibody can be covalently coupled to the stationary phase and act as the “receptor” for the antigen to be purified. Alternatively, the antigen, or parts thereof, can be attached to the stationary phase for the purification of the antibody. Advantages of immunoaffinity chromatography are its high specificity and the combination of concentration and purification in one step.
A disadvantage associated with immunoaffinity methods is the sometimes very strong antibody–anti-gen binding. This requires harsh conditions during elution of the ligand. Under such conditions, sensitive ligands could be harmed (for example, by denatura-tion of the protein to be purified). This can be alleviated by (1) the selection of antibodies and environmental conditions with high specificity and sufficient affinity to induce an antibody ligand interaction, while the antigen can be released under mild conditions (Jones, 1990); (2) the use of tandem columns to change the physical conditions (e.g., pH and ionic strength) to more physiological conditions;
(3) the use of a recipient solution into which the product is eluted. Another concern is disruption of the covalent bond linking the “receptor” to the matrix. This would result in elution of the entire complex. Therefore, in practice, a further purification step after affinity chromatography as well as an appropriate detection assay (e.g., ELISA) is almost always neces-sary. On the other hand, improved coupling chemistry that is less susceptible to hydrolysis has been developed to prevent leaching (Knight, 1990).
Scale-up of immunoaffinity chromatography is often hampered by the relatively large quantity of the specific “receptor” (either the antigen or the antibody) that is required and the lack of commercially avail-able, ready-to-use matrices.
Examples of proteins of potential therapeutic value that have been purified using immunoaffinity chromatography are interferons, urokinase, factor VIII:C, erythropoietin, interleukin-2, human factor X, and recombinant tissue plasminogen activator.
Hydrophobic Interaction Chromatography
Under physiological conditions most hydrophobic amino acid residues are located inside the protein core and only a small fraction of hydrophobic amino acids is exposed on the “surface” of a protein. Their exposure is suppressed because of the presence of hydrophilic amino acids that attract large clusters ofwater molecules and form a “shield”. High salt concentrations reduce the hydration of a protein and the surface-exposed hydrophobic amino acid residues become more accessible. Hydrophobic interaction chromatography (HIC) is based on non-covalent and non-electrostatic interactions between proteins and the stationary phase. HIC is a mild technique, usually yielding high recoveries of proteins that are not damaged, are folded correctly and are separated from contaminants that are structurally related. HIC is ideally placed in the purification scheme after ion-exchange chromatography, where the protein usually is released in high ionic strength elution media (Heng and Glatz, 1993).
Gel Permeation Chromatography
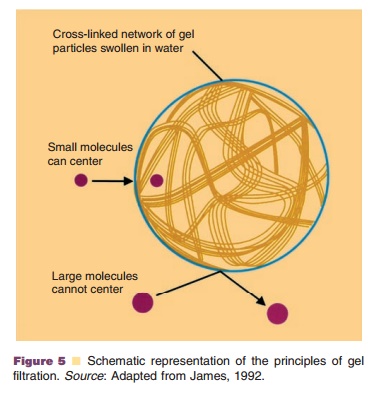
Gel-permeation or size-exclusion chromatography, also known as gel filtration, separates proteins according to their shape and size (Fig. 5). Inert gels with narrow pore-size distributions in the size range of proteins are available. These gels are packed into a column and the protein mixture is then loaded on top of the column and the proteins diffuse into the gel. The smaller the protein, the more volume it will have available in which to disperse. Molecules that are larger than the largest pores are not able to penetrate the gel beads and will therefore stay in the void volume of the column. When a continuous flow of buffer passes through the column, the larger proteins will elute first and the smallest molecules last. Gel permeation chromatography is a good alternative to diafiltration for buffer exchange at almost any purification stage, and it is often used in laboratory design. At production scale, the use of this technique is usually limited, because it requires relatively small sample volumes on a large column (up to one-third of the column volume in the case of “buffer exchange”). It is therefore best avoided or used late in the purification process when the protein is available in a highly concentrated form. Gel filtration is very commonly used as the final step in the purification to bring proteins in the appropriate buffer used in the final formulation. In this application, its use has little if no effect on the product purity characteristics.
Expanded Beds
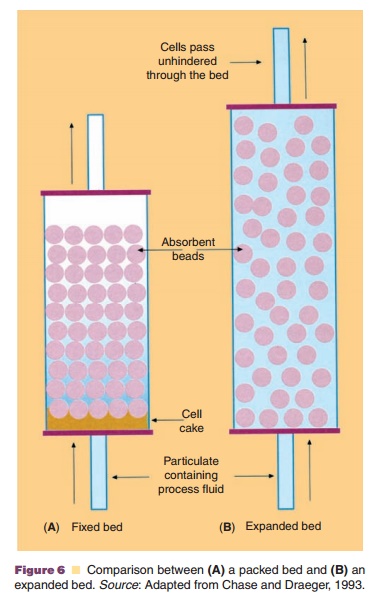
As mentioned before, purification schemes are based on multistep protocols. This not only adds greatly to the overall production costs, but also can result in significant loss of product. Therefore, there still is an interest in the development of new methods for simplifying the purification process. Adsorption techniques are popular methods for the recovery of proteins, and the conventional operating format for preparative separations is a packed column (or fixed bed) of adsorbent. Particulate material, however, can be trapped near the bed, which results in an increase in the pressure drop across the bed and eventually in clogging of the column. This can be avoided by the use of pre-column filters (0.2 mm) to save the column integrity. Another solution to this problem may be the use of expanded beds (Chase and Draeger, 1993; Fulton, 1994), also called fluidized beds (Fig. 6). In principle, the use of expanded beds enables clarifica-tion, concentration and purification to be achieved in a single step. The concept is to employ a particulate solid-phase adsorbent in an open bed with upward liquid flow. The hydrodynamic drag around the particles tends to lift them upwards, which is counter-acted by gravity because of a density difference between the particles and the liquid phase. The particles remain suspended if particle diameter, particle density, liquid viscosity and liquid density are properly balanced by choosing the correct flow rate. The expanded bed allows particles (cells) to pass through, whereas molecules in solution are selectively retained (for example, by the use of ion-exchange or affinity adsorbents) on the adsorbent particles. Feedstocks can be applied to the bed without prior removal of particulate material by centrifugation or filtration, thus reducing process time and costs. Fluidized beds have been used previously for the industrial scale recovery of antibiotics such as streptomycin and novobiocin (Chase, 1994; Fulton, 1994). Stable, expanded beds can be obtained using simple equipment adapted from that used for conventional, packed bed adsorption and chromatography processes. Ion-exchange adsorbents are likely to be chosen for such separations.
Related Topics