Chapter: Pharmaceutical Biotechnology: Fundamentals and Applications : Production and Downstream Processing of Biotech Compounds
Contaminants - Production of Biotech Compounds
Contaminants
Quality is usually measured in terms of product purity and product consistency (reproducibility). An important consideration in the development process of the purification scheme is the ultimate purity required. For pharmaceutical applications, product purity mostly is >=99% when used as a parenteral (Berthold and Walter, 1994; EMEA 1999b).
Purification processes should yield potent proteins with well-defined characteristics for human use from which “all” contaminants have been removed. The purity of the drug protein in the final product will therefore largely depend upon the purification technology applied.
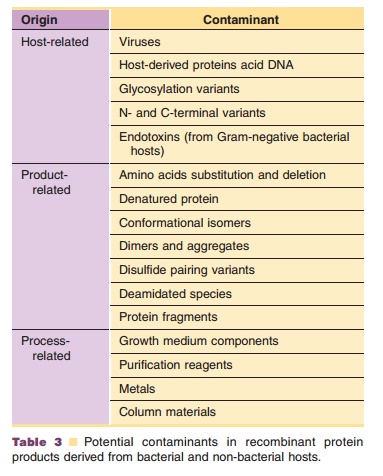
Table 3 lists potential contaminants that may be present in recombinant protein products from bacterial and non-bacterial sources. These contaminants can be either host-related, process-related or productrelated. In the following sections special attention is paid to the detection and elimination of contamination by viruses, bacteria, cellular DNA, and undesired proteins.
cultures and, therefore, of the final product produced by the cells (Arathoon and Birch, 1986). If present, their concentration in the purified product will be very low and it will be difficult to detect them. Although viruses such as retrovirus (type B) can be visualized under an electron microscope, a highly sensitive in vitro assay for their presence is lacking (Liptrot and Gull, 1991). The risks of some viruses (e.g., hepatitis virus) are known (Marcus-Sekura, 1991; Walter et al., 1991), but there are other viruses whose risks can not be properly judged because of lack of solid experimental data. Some virus infections, such as parvovirus, can have long latent periods before their clinical effects show up. Long-term effects of introducing viruses into a patient treated with a recombinant protein should not be overlooked. Therefore, it is required that parenterals are free from viruses. The specific virus testing regime required will depend on the cell type used for production (Lo¨wer, 1990; Minor, 1994).
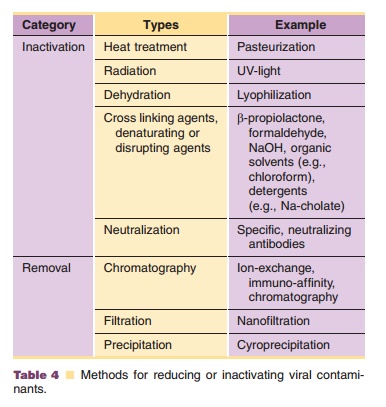
Viruses can be introduced by nutrients or they are generated by an infected production cell line. The most frequent source of virus introduction is animal serum. In addition, animal serum can introduce other unwanted agents such as bacteria, mycoplasmas, prions, fungi and endotoxins. It should be clear that appropriate screening of cell banks and growth medium constituents for viruses and other adventi-tious agents should be strictly regulated and super-vised (Walter et al., 1991; FDA 1993; EMEA, 1997; WHO Technical Report Series 823). A validated method to remove possible viral contaminants is mandatory for licensing of all therapeutics derived from mammalian cells or transgenic animals (White et al., 1991; Minor 1994). Viruses can be inactivated by physical and chemical treatment of the product. Heat, irradiation, sonication, extreme pH, detergents, sol-vents, and certain disinfectants can inactivate viruses. These procedures can be harmful to the product as well and should therefore be carefully evaluated and validated (White et al., 1991; Walter et al., 1992; Minor, 1994; EMEA, 1997). Removal of viruses by nanofiltra-tion is an elegant and effective technique and the validation aspects of this technology are extremely well described in the PDA technical report 41, 2005. Filtration through 15 nm membranes can remove even smallest non-enveloped viruses like bovine parvo-viruses (Burnouf-Radosevich et al., 1994; Maerz et al., 1996). A number of methods for reducing or inactivat-ing viral contaminants are mentioned in Table 4 (Burnouf et al., 1989; Horowitz et al., 1991; Perret et al., 1991; Horowitz et al., 1994).
Bacteria
Unwanted bacterial contamination may be a problem for cells in culture. Usually the size of bacteria allows simple sterile filtration for adequate removal. In order to further prevent bacterial contamination during production, the raw materials used have to be sterilized and the products are manufactured under strict aseptic conditions. Production most often takes place in so-called “clean rooms” in which the chances of environmental derived contamination is reduced through careful control of the environment, for example filtration of air. Additionally, antibiotic agents can be added to the culture media in some cases, but have to be removed further downstream in the purification process. The use of beta-lactam antibiotics such as penicillin is however strictly prohibited due to over-sensitivity of some individuals to these compounds. Because of the persistence of antibiotic residues, which are difficult to eliminate from the product, appropriately designed production plants and extensive quality control systems for added reagents (medium, serum, enzymes, etc.) permitting antibiotic-free operation are preferable.
Pyrogens (usually endotoxins of gram-negative bacteria) are potentially hazardous substances. Humans are sensitive to pyrogen contam-ination at very low concentrations (picograms per mL). Pyrogens may elicit a strong fever response and can even be fatal. Simple sterile filtration does not remove pyrogens. Removal is complicated further because pyrogens vary in size and chemical composi-tion. However, sensitive tests to detect and quantify pyrogens are commercially available. Purification schemes usually contain at least one step of ion-exchange chromatography (anionic exchange material) to remove the negatively charged endotoxins (Nolan, 1975; Berthold and Walter, 1994). Furthermore, materi-als used in process such as glassware are typically subjected to a depyrogenation step prior to use often by the use of elevated temperatures (180oC) in a dry heat oven.
Related Topics