Chapter: Medical Surgical Nursing: Management of Patients With Structural, Infectious, and Inflammatory Cardiac Disorders
Cardiomyopathies
Cardiomyopathies
Cardiomyopathy is
a heart muscle disease associated with cardiacdysfunction. It is classified
according to the structural and func-tional abnormalities of the heart muscle:
dilated cardiomyopathy (DCM), hypertrophic cardiomyopathy (HCM), restrictive
orconstrictive cardiomyopathy, arrhythmogenic right ventricular
cardiomyopathy (ARVC), and
unclassified cardiomyopathy
(Richardson et al., 1996). Ischemic
cardiomyopathy is a term frequently used to describe an enlarged heart
caused by coronary artery disease, which is usually accompanied by heart
failure . Regardless of the category and the cause, cardio- myopathy may lead
to severe heart failure, lethal dysrhythmias,and death. Cardiomyopathy causes
more than 27,000 deaths each year in the United States (American Heart
Association, 2001). The mortality rate is highest for African Americans and the
elderly (American Heart Association, 2001).
Pathophysiology
The
pathophysiology of all cardiomyopathies is a series of pro-gressive events that
culminate in impaired cardiac output. De-creased stroke volume stimulates the
sympathetic nervous system and the renin-angiotensin-aldosterone response,
resulting in in-creased systemic vascular resistance and increased sodium and
fluid retention, which places an increased workload on the heart. These
alterations can lead to heart failure .
DILATED CARDIOMYOPATHY
DCM
is the most common form of cardiomyopathy, with an in-cidence of 5 to 8 cases
per 100,000 people per year and increas-ing (Braunwald et al., 2001). DCM
occurs more often in men and African Americans, who also experience higher
mortality rates (Braunwald et al., 2001). DCM is distinguished by signifi-cant
dilation of the ventricles (Fig. 29-8) without significant concomitant
hypertrophy (ie, increased muscle wall thickness) and systolic dysfunction. DCM
was formerly named congestivecardiomyopathy,
but DCM may exist without signs and symp-toms of congestion.
Microscopic
examination of the muscle tissue shows dimin-ished contractile elements of the
muscle fibers and diffuse necrosis of myocardial cells. The result is poor
systolic function. These structural changes decrease the amount of blood
ejected from the ventricle with systole, increasing the amount of blood
remaining in the ventricle after contraction. Less blood is then able to enter
the ventricle during diastole, increasing end-diastolic pressure and eventually
increasing pulmonary pressures. Altered valve function can result from the
enlarged stretched ventricle, usually resulting in regurgitation. Embolic
events caused by ventricular and atrial thrombi as a result of the poor blood
flow through the ventricle may also occur. More than 75 conditions and diseases
may cause DCM, including pregnancy, heavy alcohol intake, and viral infection
(eg, influenza). When the causative factor cannot be identified, the term used
is idiopathic DCM. Idiopathic DCM
accounts for approximately 25% of all heart failure cases (Braunwald et al.,
2001). Early diagnosis and treatment can pre-vent or delay significant symptoms
and sudden death from DCM. Echocardiography and ECG are used to diagnose DCM
and should be conducted for all first-degree relatives (eg, parents, siblings,
children) of patients with DCM (Braunwald et al., 2001).
HYPERTROPHIC CARDIOMYOPATHY
In HCM, the heart muscle increases in size and mass, especially along the septum (see Fig. 29-8). The increased thickness of the heart muscle reduces the size of the ventricular cavities and causes the ventricles to take a longer time to relax, making it more diffi-cult for the ventricles to fill with blood during the first part of di-astole and making them more dependent on atrial contraction for filling.
The increased septal size may misalign the papillary mus-cles so that the
septum and mitral valve obstruct the flow of blood from the left ventricle into
the aorta during ventricular contrac-tion. Hence, HCM may be obstructive or
nonobstructive. Because of the structural changes, HCM had also been called
idiopathic hypertrophic subaortic stenosis (IHSS) or asymmetric septal
hyper-trophy (ASH). Structural changes may also result in a smaller than normal
ventricular cavity and a higher velocity flow of blood out of the left ventricle
into the aorta, which may be de-tected by echocardiography (Braunwald et al.,
2001). HCM may cause significant diastolic dysfunction, but systolic function
can be normal or high, resulting in a higher than normal ejection fraction.
Because
HCM is a genetic disease, family members are ob-served closely for signs and
symptoms indicating development of the disease (Fuster et al., 2001). HCM is
rare, occurring in men, women, and children (often detected after puberty)
(Oakley, 1997) with an estimated prevalence rate of 0.05% to 0.2% (Berul
Zevitz, 2002). It may also be idiopathic (ie, no cause can be found).
RESTRICTIVE CARDIOMYOPATHY
Restrictive
cardiomyopathy (RCM) is characterized by diastolic dysfunction caused by rigid
ventricular walls that impair ventric-ular stretch and diastolic filling (see
Fig. 29-8). Systolic function is usually normal. Because RCM is the least
common cardio-myopathy, representing approximately 5% of pediatric
cardiomy-opathies, its pathogenesis is the least understood (Shaddy, 2001).
Restrictive cardiomyopathy can be associated with amyloidosis (in which
amyloid, a protein substance, is deposited within thecells) and other such
infiltrative diseases. However, the cause is unknown in most cases (ie,
idiopathic).
ARRHYTHMOGENIC RIGHT VENTRICULAR CARDIOMYOPATHY
ARVC
occurs when the myocardium of the right ventricle is pro-gressively infiltrated
and replaced by fibrous scar and adipose tis-sue. Initially, only localized
areas of the right ventricle are affected, but as the disease progresses, the
entire heart is affected. Eventually, the right ventricle dilates and develops
poor contractility, right ventricular wall abnormalities, and dysrhythmias. The
prevalence of ARVC is unknown because many cases are not recognized. ARVC should
be suspected in patients with ventricular tachy-cardia originating in the right
ventricle (ie, a left bundle branch block configuration on ECG) or sudden
death, especially among previously symptom-free athletes (McRae et al., 2001).
The dis-ease may be genetic (ie, autosomal dominant) (Richardson et al., 1996).
Family members should be screened for the disease with a 12-lead ECG, Holter
monitor, and echocardiography.
UNCLASSIFIED CARDIOMYOPATHIES
Unclassified
cardiomyopathies are different from or have char-acteristics of more than one
of the previously described cardio-myopathies. Examples of unclassified
cardiomyopathies include fibroelastosis, noncompacted myocardium, systolic
dysfunc-tion with minimal dilation, and mitochondrial involvement (Richardson
et al., 1996).
Clinical Manifestations
The
patient may have cardiomyopathy but remain stable and without symptoms for many
years. As the disease progresses, so do symptoms. Frequently, dilated and
restrictive cardiomyopathy are first diagnosed when the patient presents with
signs and symp-toms of heart failure (eg, dyspnea on exertion, fatigue).
Patients with cardiomyopathy may also report paroxysmal nocturnal dys-pnea,
cough (especially with exertion), and orthopnea, which may lead to a
misdiagnosis of bronchitis or pneumonia. Other symp-toms include fluid
retention, peripheral edema, and nausea, which is caused by poor perfusion of
the gastrointestinal system. The pa-tient may experience chest pain,
palpitations, dizziness, nausea, and syncope with exertion. However, with HCM,
cardiac arrest (ie, sudden cardiac death) may be the initial manifestation in
young people, including athletes (Spirito et al., 2000).
Assessment and Diagnostic Findings
Physical
examination in the early stage may reveal tachycardia and extra heart sounds.
With disease progression, examination also reveals signs and symptoms of heart
failure (eg, crackles on pul-monary auscultation, jugular vein distention,
pitting edema of dependent body parts, enlarged liver).
Diagnosis
is usually made from findings disclosed by the pa-tient history and by ruling
out other causes of heart failure, such as myocardial infarction. The
echocardiogram is one of the most helpful diagnostic tools because the
structure and function of the ventricles can be observed easily. ECG
demonstrates dysrhyth-mias and changes consistent with left ventricular
hypertrophy. The chest x-ray film reveals heart enlargement and possibly
pul-monary congestion. Cardiac catheterization is sometimes used to rule out
coronary artery disease as a causative factor. An en-domyocardial biopsy may be
performed to analyze myocardial tissue cells.
Medical Management
Medical
management is directed toward determining and man-aging possible underlying or
precipitating causes; correcting the heart failure with medications, a
low-sodium diet, and an exercise-rest regimen ; and controlling dysrhythmias
with antiarrhythmic medications and possibly with an implanted elec-tronic
device, such as an implantable cardioverter-defibrillator . If patients exhibit
signs and symptoms of conges-tion, their fluid intake may be limited to 2
liters each day. The person with HCM may also have to limit physical activity
to avoid a life-threatening dysrhythmia. A pacemaker may be implanted to alter
the electrical stimulation of the muscle and prevent the forceful hyperdynamic
contractions that occur with HCM.
SURGICAL MANAGEMENT
When
heart failure progresses and medical treatment is no longer effective, surgical
intervention, including heart transplantation, is considered. However, because
of the limited number of organ donors, many patients die waiting for
transplantation. In some cases, a left ventricular assist device (LVAD) is
implanted to sup-port the failing heart until a suitable donor heart becomes avail-able.
Left Ventricular Outflow Tract Surgery.
When a patient withHCM becomes symptomatic despite medical therapy
and a dif-ference in pressure of 50 mm Hg or more exists between the left
ventricle and the aorta, surgery is considered. The most common procedure is a
myectomy (sometimes referred to as a myotomy-myectomy), in which some of the
heart tissue is excised. Septal tissue approximately 1 cm wide and deep is cut
from the enlarged septum below the aortic valve. The length of septum removed
de-pends on the degree of obstruction caused by the hypertrophied muscle.
Instead
of a septal myectomy, the surgeon may open the left ventricular outflow tract
to the aortic valve by removing the mi-tral valve, chordae, and papillary
muscles. The mitral valve then is replaced with a low-profile disk valve. The
space taken up by the mitral valve is substantially reduced by the prosthetic
valve compared with the patient’s own valve, chordae, and papillary muscles,
allowing blood to move around the enlarged septum to the aortic valve in the
area that the mitral valve once occupied. The primary complication of both
procedures is dysrhythmia; additional complications are postoperative surgical
complications such as pain, ineffective airway clearance, deep vein thrombosis,
risk for infection, and delayed surgical recovery.
Heart Transplantation.
The first human-to-human heart trans-plant was performed in 1967.
Since then, transplant procedures, equipment, and medications have continued to
improve. Since 1983, when cyclosporine became available, heart transplantation
has become a therapeutic option for patients with end-stage heart disease.
Cyclosporine (Neoral, Sandimmune, SangCya) is an immunosuppressant that greatly
decreases the body’s rejection of foreign proteins, such as transplanted
organs. Unfortunately, cyclosporine also decreases the body’s ability to resist
infections, and a satisfactory balance must be achieved between suppressing
rejection and avoiding infection.
Cardiomyopathy,
ischemic heart disease, valvular disease, rejection of previously transplanted
hearts, and congenital heart disease are the most common indications for
transplantation (Becker & Petlin, 1999; Rourke et al., 1999). A typical
candidatehas severe symptoms uncontrolled by medical therapy, no other surgical
options, and a prognosis of less than 12 months to live. A multidisciplinary
team screens the candidate before recommend-ing the transplantation procedure.
The person’s age, pulmonary status, other chronic health conditions, psychosocial
status, fam-ily support, infections, history of other transplantations,
compli-ance, and current health status are considered in the screening.
When
a donor heart becomes available, a computer generates a list of potential
recipients on the basis of ABO blood group compatibility, the sizes of the
donor and the potential recipient, and the geographic locations of the donor
and potential recipient; distance is a variable because postoperative function
depends on the heart being implanted within 6 hours of harvest from the donor.
Some patients are candidates for more than one organ transplant: heart-lung,
heart-pancreas, heart-kidney, heart-liver.
Transplantation Techniques.
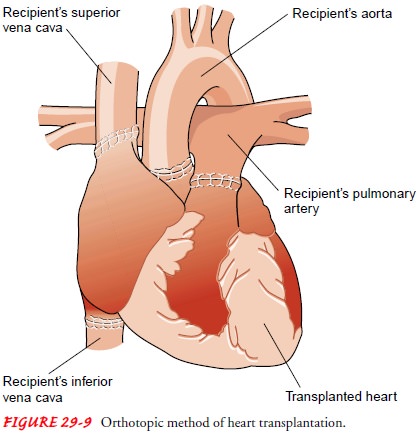
Orthotopic
transplantationis themost common surgical procedure for cardiac
transplantation (Fig. 29-9). The recipient’s heart is removed, and the donor
heart is implanted at the vena cava and pulmonary veins. Some sur-geons still
prefer to remove the recipient’s heart leaving a portion of the recipient’s
atria (with the vena cava and pulmonary veins) in place. The donor heart, which
usually has been preserved in ice, is prepared for implant by cutting away a
small section of the atria that corresponds with the sections of the
recipient’s heart that were left in place. The donor heart is implanted by
suturing the donor atria to the residual atrial tissue of the recipient’s
heart. Both techniques then connect the recipient’s pulmonary artery and aorta
to those of the donor heart.
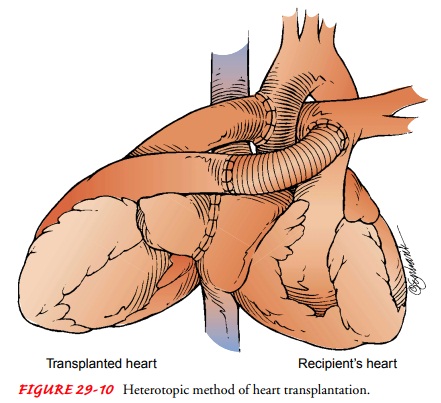
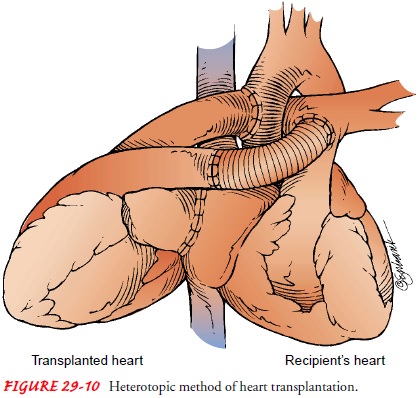
Heterotopic transplantation is
less commonly performed(Fig. 29-10). The donor heart is placed to the right and
slightly anterior to the recipient’s heart; the recipient’s heart is not
removed. Initially, it was thought that the original heart might provide some
protection for the patient in the event that the transplanted heart was
rejected. Although the protective effect has not been proved, other reasons for
retaining the original heart have been identified: a small donor heart or
pulmonary hyper-tension (Becker & Petlin, 1999; Kadner et al., 2000).
The
transplanted heart has no nerve connections with the re-cipient’s body (ie,
denervated heart), and the sympathetic and vagus nerves do not affect the
transplanted heart. The resting rate of the transplanted heart is approximately
70 to 90 beats per minute, but it increases gradually if catecholamines are in
the cir-culation. Patients must gradually increase and decrease their ex-ercise
(ie, extended warm-up and cool-down periods), because 20 to 30 minutes may be
required to achieve the desired heart rate. Atropine does not increase the
heart rate of these patients.
Postoperative Course.
Heart
transplant patients are constantlybalancing the risk of rejection with the risk
of infection. They must comply with a complex regimen of diet, medications,
ac-tivity, follow-up laboratory studies, biopsies (to diagnose rejection), and
clinic visits. Most commonly, patients receive cyclosporine or tacrolimus
(FK506, Prograf), azathioprine (Imuran) or myco-phenolate mofetil (CellCept),
and corticosteroids (ie, prednisone) to minimize rejection.
In addition to rejection and
infection, complications may in-clude accelerated atherosclerosis of the
coronary arteries (ie, cardiac allograft vasculopathy [CAV ]or accelerated
graft atherosclerosis [AGA]). Although the cause is unknown, the disease is
believed to be immunologically mediated (Augustine, 2000; Rourke et al., 1999).
Hypertension may be experienced by patients taking cy-closporine or tacrolimus;
the cause has not been identified. Osteoporosis frequently occurs as a side
effect of the anti-rejection medications and pretransplantation dietary
insufficiency and med-ications. Posttransplantation lymphoproliferative disease
and can-cer of the skin and lips are the most common malignancies after
transplantation, possibly caused by immunosuppression. Weight gain, obesity,
diabetes, dyslipidemias (eg, hypercholesterolemia), hypotension, renal failure,
and central nervous system, respiratory, and gastrointestinal disturbances may
be caused by the cortico-steroids or other immunosuppressants. Other
complications are immunosuppressant medication toxicities and responses to the
psychosocial stresses imposed by organ transplantation. Patients may experience
guilt that someone died for them to live, have anxiety about the new heart,
experience depression or fear when rejection is identified, or have difficulty
with family role changes before and after transplantation (Augustine, 2000;
Becker &Petlin, 1999; Braunwald et al., 2001; Fuster et al., 2001; Rourke
et al., 1999).
The
1-year survival rate for patients with transplanted hearts is approximately 80%
to 90%; the 5-year survival rate is ap-proximately 60% to 70% (Augustine, 2000;
Becker & Petlin, 1999; Braunwald et al., 2001; Fuster et al., 2001; Rourke
et al., 1999).
Mechanical Assist Devices and Total Artificial Hearts.
The useof cardiopulmonary bypass for
cardiovascular surgery and the possibility of performing heart transplantation
for end-stage car-diac disease have increased the need for mechanical assist
devices. Patients who cannot be weaned from cardiopulmonary bypass or patients
in cardiogenic shock may benefit from a period of me-chanical heart assistance.
The most commonly used device is the intra-aortic balloon pump . This pump
decreases the work of the heart during contraction but does not perform the
actual work of the heart.
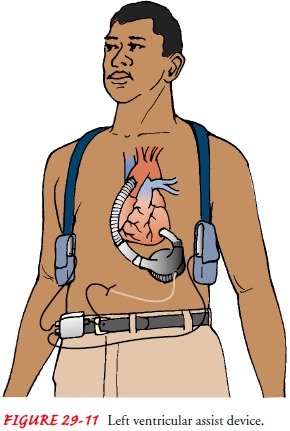
Ventricular Assist Devices.
More complex devices that actuallyperform some or all of the
pumping function for the heart also are being used. These more sophisticated ventricular assist devices (VADs) (Fig.
29-11) can circulate as much blood per minute as the patient’s heart, if not
more. Each ventricular assist device is used to support one ventricle. Some
ventricular assist devices can be combined with an oxygenator; the combination
is called extra-corporeal membrane oxygenation (ECMO). The oxygenator–
ventricular assist device combination is used for the patient whose heart
cannot pump adequate blood through the lungs or the body.
There
are three basic types of devices: centrifugal, pneumatic, and electric or
electromagnetic. Centrifugal VADs are external, nonpulsatile, cone-shaped
devices with internal mechanisms that spin rapidly, creating a vortex
(tornado-like action) that pulls blood from a large vein into the pump and then
pushes it back into a large artery. Pneumatic VADs are external or implanted
pulsatile devices with a flexible reservoir housed in a rigid exterior. The
reservoir usually fills with blood drained from the pa-tient’s atrium or
ventricle. The VAD then forces pressurized air into the rigid housing,
compressing the reservoir and returning the blood to the patient’s circulation,
usually into the aorta. Elec-tric or electromagnetic VADs are similar to the
pneumatic VADs, but instead of pressurized air, one or more flat metal plates
are pushed against the reservoir to return the blood to the patient’s
circulation.
Total Artificial Hearts.
Total artificial heartsare designed to re-place both ventricles. Some require the removal
of the patient’s heart to implant the total artificial heart; others do not.
All of these devices are experimental. Although there has been some short-term
success, the long-term results have been disappointing. Researchers hope to
develop a device that can be permanently im-planted and that will eliminate the
need for donated human heart transplantation for the treatment of end-stage
cardiac disease (Braunwald et al., 2001; Chillcott et al., 1998; Fuster et al.,
2001; Rose et al., 1999; Schakenbach, 2001).
Most
VADs and total artificial hearts are temporary treat-ments while the patient’s
own heart recovers or until a donor heart becomes available for transplantation
(ie, “bridge to trans-plant”). Some devices are being investigated for permanent
use. Bleeding disorders, hemorrhage, thrombus, emboli, hemolysis, infection,
renal failure, right heart failure, multisystem failure, and mechanical failure
are some of the complications of VADs and total artificial hearts (Braunwald et
al., 2001; Duke & Perna, 1999; Schakenbach, 2001; Scherr et al., 1999). The
nursing care for these patients focuses on assessing for and minimizing these
complications and involves providing emotional support and education about the
mechanical assist device.
Related Topics