Chapter: Basic & Clinical Pharmacology : Vasoactive Peptides
Biosynthesis of Angiotensin
BIOSYNTHESIS OF ANGIOTENSIN
The
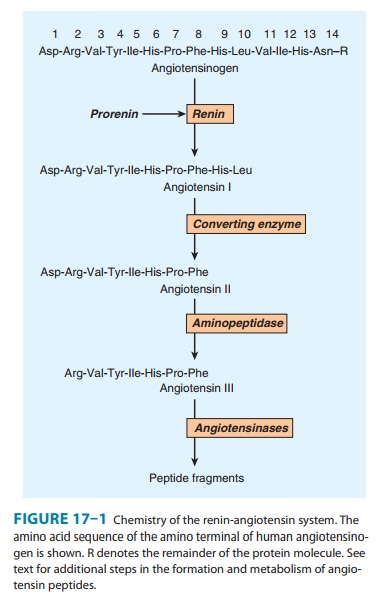
pathway for the formation and metabolism of angiotensin (ANG II) is summarized
in Figure 17–1. The principal steps include enzymatic cleavage of angiotensin I
(ANG I) from angiotensinogen by renin, conversion of ANG I to ANG II by converting
enzyme, and degradation of ANG II by several peptidases.
Renin
Renin
is an aspartyl protease enzyme that specifically catalyzes the hydrolytic
release of the decapeptide ANG I from angiotensinogen. It is synthesized as a
prepromolecule that is processed to prorenin, which has poorly understood
actions , and then to active renin, a glycoprotein consisting of 340 amino
acids.
Renin
in the circulation originates in the kidneys. Enzymes with renin-like activity
are present in several extrarenal tissues, including blood vessels, uterus,
salivary glands, and adrenal cortex, but no physiologic role for these enzymes
has been established. Within the kidney, renin is synthesized and stored in the
juxta-glomerular apparatus of the nephron. Specialized granular cells called
juxtaglomerular cells are the site of synthesis, storage, and release of renin.
The macula densa is a specialized segment of the nephron that is closely
associated with the vascular components of the juxtaglomerular apparatus. The
vascular and tubular compo-nents of the juxtaglomerular apparatus, including
the juxtaglom-erular cells, are innervated by noradrenergic neurons.
Control of Renin Release
The rate at which renin is released by the kidney is the primary determinant of activity of the renin-angiotensin system.
Active renin
is released by exocytosis immediately upon stimulation of the juxtaglomerular
apparatus. Prorenin is released constitutively, usually at a rate higher than
that of active renin, thus accounting for the fact that prorenin can constitute
80–90% of the total renin in the circulation. The significance of circulating
prorenin is dis-cussed at the end of this section. Active renin release is
controlled by a variety of factors, including a renal vascular receptor, the
macula densa, the sympathetic nervous system, and ANG II.
A. Macula Densa
Renin
release is controlled in part by the macula densa, a structure that has a close
anatomic association with the afferent arteriole. The initial step involves the
detection of some function of NaCl concentration in, or delivery to, the distal
tubule, possibly by the Na+/K+/2Cl–
cotransporter. The macula densa then signals changes in renin release by the
juxtaglomerular cells such that there is an inverse relationship between NaCl
delivery or concentration and renin release. Potential candidates for signal
transmission include prostaglandin E2 (PGE2) and nitric
oxide, which stimulate renin release, and adenosine, which inhibits it.
B. Renal Baroreceptor
The
renal baroreceptor mediates an inverse relationship between renal artery
pressure and renin release. The mechanism is not completely understood but it
appears that the juxtaglomerular cells are sensitive to stretch and that
increased stretch results in decreased renin release. The decrease may result
from influx of calcium which, somewhat paradoxically, inhibits renin release.
The paracrine factors PGE2, nitric oxide, and adenosine have also
been implicated in the baroreceptor control of renin release.
C. Sympathetic Nervous System
Norepinephrine
released from renal sympathetic nerves stimu-lates renin release indirectly by α-adrenergic activation
of the renal baroreceptor and macula densa mechanisms, and directly by an
action on the juxtaglomerular cells. In humans, the direct effect is mediated
by β1 adrenoceptors. Through this mecha-nism, reflex activation of
the sympathetic nervous system by hypotension or hypovolemia leads to
activation of the renin-angiotensin system.
D. Angiotensin
Angiotensin
II inhibits renin release. The inhibition results from increased blood pressure
acting by way of the renal baroreceptor and macula densa mechanisms, and from a
direct action of the peptide on the juxtaglomerular cells. The direct
inhibition is mediated by increased intracellular Ca2+ concentration
and forms the basis of a short-loop negative feedback mechanism controlling
renin release. Interruption of this feedback with drugs that inhibit the
renin-angiotensin system results in
stimulation of renin release.
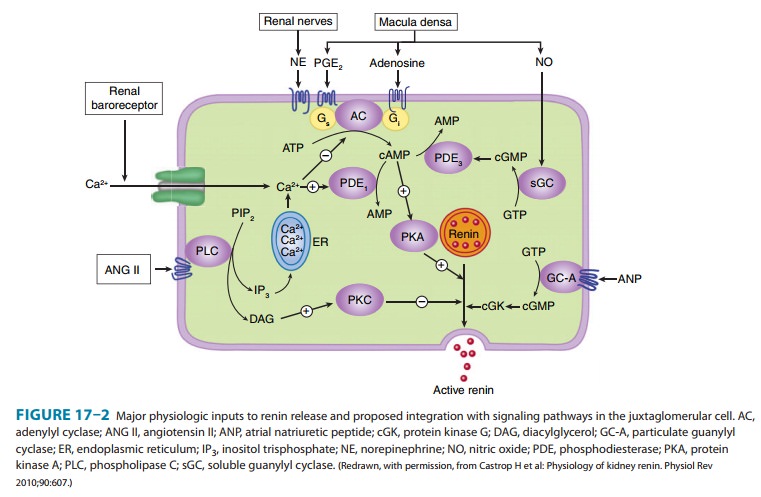
E. Intracellular Signaling Pathways
The
release of renin by the juxtaglomerular cells is controlled by interplay among
three intracellular messengers: cAMP, cyclic guanosine monophosphate (cGMP),
and free cytosolic Ca2+ con-centration (Figure 17–2). cAMP plays a
major role; maneuvers that increase cAMP levels, including activation of
adenylyl cyclase, inhibition of cAMP phosphodiesterases, and administration of
cAMP analogs, increase renin release. Increases in Ca2+ can result
from increased entry of extracellular Ca2+ or mobilization of Ca2+
from intracellular stores, while increases in cGMP levels can result from
activation of soluble or particulate guanylyl cyclase. Ca2+ and cGMP
appear to alter renin release indirectly, primarily by chang-ing cAMP levels.
F. Pharmacologic Alteration of Renin Release
The
release of renin is altered by a wide variety of pharmacologic agents. Renin
release is stimulated by vasodilators (hydralazine, minoxidil, nitroprusside), β-adrenoceptor
agonists, α-adrenoceptor
antagonists, phosphodiesterase inhibitors (eg, theophylline, milri-none,
rolipram), and most diuretics and anesthetics. This stimula-tion can be
accounted for by the control mechanisms just described. Drugs that inhibit
renin release are discussed below.
Many of the peptides reviewed also alter renin release. Release is stimulated by adrenomedullin, bradykinin, and calcitonin gene-related peptide, and inhibited by atrial natriuretic peptide, endothelin, substance P, and vasopressin.
Angiotensinogen
Angiotensinogen
is the circulating protein substrate from which renin cleaves ANG I. It is
synthesized in the liver. Human angio-tensinogen is a glycoprotein with a
molecular weight of approxi-mately 57,000. The 14 amino acids at the amino
terminal of the molecule are shown in Figure 17–1. In humans, the
concentra-tion of angiotensinogen in the circulation is less than the Km
of the renin-angiotensinogen reaction and is therefore an impor-tant
determinant of the rate of formation of angiotensin.The production of
angiotensinogen is increased by corticosteroids, estrogens, thyroid hormones,
and ANG II. It is also elevated during pregnancy and in women taking
estrogen-containing oral contracep-tives. The increased plasma angiotensinogen
concentration is thought to contribute to the hypertension that may occur in
these situations.
Angiotensin I
Although
ANG I contains the peptide sequences necessary for all of the actions of the
renin-angiotensin system, it has little or no biologic activity. Instead, it
must be converted to ANG II by con-verting enzyme (Figure 17–1). ANG I may also
be acted on by plasma or tissue aminopeptidases to form [des-Asp1]angiotensin
I; this in turn is converted to [des-Asp1]angiotensin II (commonly
known as angiotensin III) by converting enzyme.
Converting Enzyme (ACE, Peptidyl Dipeptidase, Kininase II)
Converting
enzyme is a dipeptidyl carboxypeptidase with two active sites that catalyzes
the cleavage of dipeptides from the carboxyl terminal of certain peptides. Its
most important sub-strates are ANG I, which it converts to ANG II, and
bradykinin, which it inactivates (see Kinins, below). It also cleaves
enkepha-lins and substance P, but the physiologic significance of these effects
has not been established. The action of converting enzyme is prevented by a
penultimate prolyl residue in the sub-strate, and ANG II is therefore not
hydrolyzed by converting enzyme. Converting enzyme is distributed widely in the
body. In most tissues, converting enzyme is located on the luminal sur-face of
vascular endothelial cells and is thus in close contact with the circulation.
A
homolog of converting enzyme known as ACE2 was recently found to be highly
expressed in vascular endothelial cells of the kidneys, heart, and testes.
Unlike converting enzyme, ACE2 has only one active site and functions as a
carboxypeptidase rather than a dipeptidyl carboxypeptidase. It removes a single
amino acid from the C-terminal of ANG I forming ANG 1-9 (Figure 17–3), which is
inac-tive but is converted to ANG 1-7 by ACE. ACE2 also converts ANG
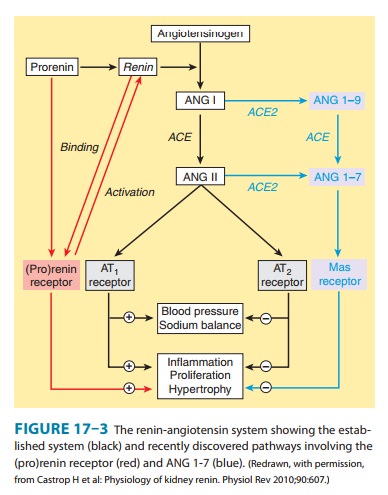
This vasodilation may serve to counteract the
vasoconstrictor activity of ANG II. ACE2 also differs from ACE in that it does
not hydrolyze bradykinin and is not inhib-ited by converting enzyme inhibitors
. Thus, the enzyme more closely resembles an angiotensinase than a converting
enzyme.
Angiotensinase
Angiotensin
II, which has a plasma half-life of 15–60 seconds, is removed rapidly from the
circulation by a variety of peptidases col-lectively referred to as
angiotensinase. It is metabolized during passage through most vascular beds (a
notable exception being the lung). Most metabolites of ANG II are biologically
inactive, but the initial product of aminopeptidase action—[des-Asp1]angiotensin
II—retains considerable biologic activity.
Related Topics