Chapter: Pharmaceutical Drug Analysis: Theory and Technique of Quantitative Analysis
Biomedical Analytical Chemistry: Enzymatic Assays
ENZYMATIC ASSAYS
A. Theory :
All colorimetric enzymatic
assays essentially involve the measurement of the activity of an ezyme under the following two circumstances, namely :
(a) When
substrate is in large excess, and
(b) When enzyme
concentration is in large excess.
A.1. Substrate Present in Large Excess :
In reality, an enzyme reaction is nothing but a special
kind of generalized reaction that
may best be expressed as follows :
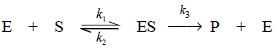 .....................(a)
.....................(a)
Where, E = Enzyme,
S = Substrate
ES =
Enzyme-substrate complex, and
P =
Product.
From Eq. (a), we have,
Rate of Product Formation = Vmax [S]/Km + [S] ...(b)
Where, Km =
(k2 + k3) / k1,
Vmax = Max. rate of reaction
Assuming, [S] to be in large excess [S] >> Km,
From Eq. (b) we
have :
Rate of Reaction = Vmax [S]/[S]
or Rate of Reaction = Vmax
...(c)
Example : In order to measure the
activity of an enzyme E, such as creatine phosphokinase (CPK), the concentration
of the substrate S, for instance creatine, should be in large excesses so that
the products measured shall be in the linear portion of the curve (Part ‘A’) in
Figure 2.5.
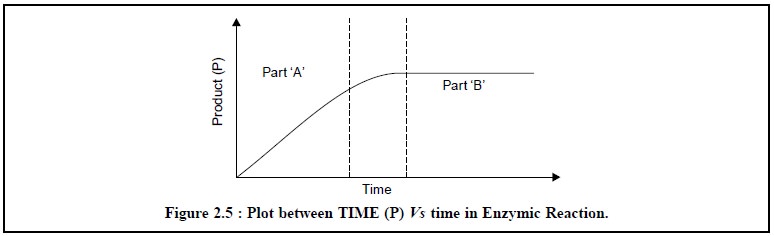
Therefore, with a view to obtaining the best results, the
two experimental parameters, namely : the temperature
(constant-temperature-water-bath) and the time (phaser) should always be kept
constant in order that the rate of reaction, as determined by the amount of
product formed, specially designates the activity of the enzyme under assay,
and devoid of the influence of any other variables on the reaction rate.
A.2. Enzyme Concentration in Large Excess
In order to analyze the quantity of substrate (S) present
in a biological sample glucose oxidase is added in excess of the actual amount
needed for the complete conversion of all the substrate to product ; and to
achieve this object the reaction is allowed to run for a fairly long duration (i.e., to complete the reaction). It can
be seen evidently in Part ‘B’ of Figure 2.5, wherein the sepecific reaction
time the substrate (S) has been consumed completely and consequently, the
concentration of the product achieves a maximum value.
1. Assay Methods
A few typical examples of colorimetric assay of enzyme
levels will be discussed briefly hereunder :
1.1. Alkaline Phosphatase (AP)
Theory :
Alkaline phosphatase is
responsible for the cleavage of O-P bonds. It is found to be relatively non-specific and this characteristic
permits the AP level to be assayed based on the fact that p-nitrophenylphosphate ion gets converted to p-nitrophenolate anion at pH 10.5; as expressed in the following
reaction.
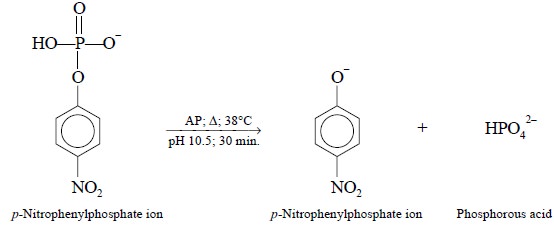
In actual practice, p-nitrophenylphosphate
is present in large excesses, and the reaction is carried out at 38°C for 30
minutes. The resulting amount of p-nitrophenolate
ion is estimated by the help of an usual standard curve employing known concentrations
of p-nitrophenolate prepared from p-nitrophenol.
Bessey-Lowry Activity :
One unit of activity may be
defined as the amount of enzyme present in
1 millilitre of serum that liberates 1 μ mol of p-nitrophenol
(0.1391 mg)* per hour at pH 10.5 after 30 minutes at 38°C.
Elimination of Interference due to Coloured Drugs
p-Nitrophenol is colourless,
whereas the phenolate ion under basic conditions is yellow in appeanace. Therefore, the elimination of
interference due to coloured drugs present in the serum is accomplished
effectively by first, measuring the
absorbance of the serum under basic conditions, and secondly, under acidic conditions. Thus we have :
Ap-nitrophenolate = Abasic – Aacidic
Profile of AP-levels
·
Normal AP-levels in adults range between 0.8 to 2.3
Bessey-Lowry units and in children between 2.8 to 6.7,
·
Increased AP-levels are observed in patients suffering
from liver diseases, hyperparathyroidism and in rickets,
·
Decreased AP-levels could be seen in patients suffering
from hypoparathyroidism and pernicious anaemia (i.e., an anaemia tending to be a fatal issue).
Interference due to Bilirubin
Bilirubin is eliminated by dializing the incubated p-nitrophenolate ion (at pH 10.5, and
maintaining at 38°C for 30 minutes) into 2-amino-2-methyl-1-propanol, without
carrying out the blank determination stated earlier.
There are a few medicinals that cause increased bilirubin
levels which ultimately enhances AP-levels ; unless and until a corrective
measure is taken in the respective procedure one may be left with false
AP-level enhancement. Some typical examples are, namely : amitriptyline,
chloropropamide, erythromycin, phenylbutazone, sulpha-drugs and tetracyclines.
Materials Required :
0.01 M p-Nitrophenol (dissolve
140 mg of p-nitrophenol in 100 ml of DW) : 1.0 ml ; 0.02 N NaOH (dissolve 160 mg in 200 ml DW) : 200 ml ; 5
ml of alkaline-buffered substrate (l M p-nitrophenylphosphate)
(dissolve 7.5 g glycine, 0.095 g anhydrous MgCl2 and 85 ml of 1 N
NaOH to 1 litre with DW ; and mixing with an equal volume of a solution
prepared by dissolving 0. l0 g of p-nitro-phenylphosphate
in 25 ml of water) ; temperature bath previously set at 38°C ; alkaline
phosphatase for unknowns (commercial source) ; working standard [dilute 0.50 ml
of a solution of p-nitrophenol (10.0
mol/ litre, 0.139 g/100 ml) to 100 ml with 0.02 N NaOH].
Procedure :
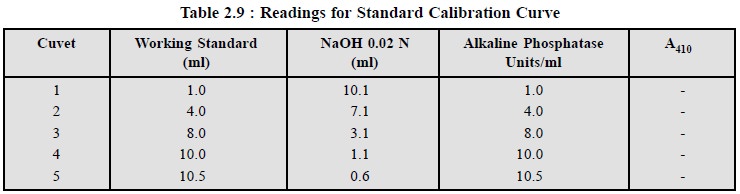
(1) First of all prepare a
standard calibration curve as per Table 2.9.
(2) Plot a graph of absorbance
A Vs units of alkaline phosphatase
per millilitre.
(3) Proceed for the assay of
AP in the serum sample sequentially as follows :
(i) Pipette 1.0
ml of alkaline—buffered substrate into each of two test tubes and keep in a
water-bath preset at 38°C,
(ii) When both
the test tubes have attained the temperature equlibrium, add 0.10 ml of serum
and water to these tubes separately. The one with water serves as a reagent
blank and is always needed per set of unknowns. Now, put the two tubes for
incubation for exactly 30 minutes period,
(iii) Enzyme
activity is arrested by adding 10.0 ml of 0.02 N NaOH to each tube. Remove them
from the water-bath and mix the contents thoroughly,
(iv) Read out
the absorbance of the unknown tube at 410 nm against the ‘reagent blank’ tube,
(v) Transfer
the contents from the cuvets to the respective test-tubes and add 0.1 ml of HCl
( −~ 11.5 N to each tube and mix the contents carefully.
This operation removes the colour developed due to p-nitrophenol,
(vi) Again read
out the absorbance of the serum sample against the reagent blank tube at 410
nm. This gives the colour due to the serum itself,
(vii) Now, the
corrected reading is achieved by subtracting the reading obtained in step (vi) from the reading in step (v). The alkaline-phosphatase activity of
the serum as Bessey-Lowery units is obtained from the calibration-curve step (i). Under these experimental parameters,
we have :
1 Bessey-Lowry Unit = 5 × 10 –8 mol of p-Nitrophenolate anion.
Thus, one unit of phosphatase activity liberated 1 μ mol of p-nitrophenol
(l μ mol = 0.1391 mg) per hour per millilitre of serum under
specified conditions.
Note : In case, a value more than 10
Bessey-Lowry Units is obtained, it is always advisable to repeat the process
either with a smaller volume of serum or a shorter incubation period, and then
finally adjust the calculations accordingly.
(4) Report the concentration of AP in units per
millilitre.
1.2. Lactate Dehydrogenase (LDH)
Theory :
The method of LDH assay is
based on kinetic analysis. In a kinetic enzymatic assay a unit of enzyme activity is defined as ‘the quantity of enzyme that brings about a
certain absorbance increase in 30
seconds or 1 minute at a fixed temperature (for instance 25 ± 0.2°C) ’.
The kinetic assay of LDH is
based on the conversion of lactic acid to pyruvic acid, in the presence of
nicotinamide adenine dinucleotide (NAD), and is closely monitored at intervals
of 30 seconds or 1 minute by measuring the increase in absorbance at 340 nm. In
this particular instance lactic acid available in an excess to ensure that the
increase in pyruvic acid is linear with time, i.e., directly proportional to time. The reaction involved may be
expressed as follows :
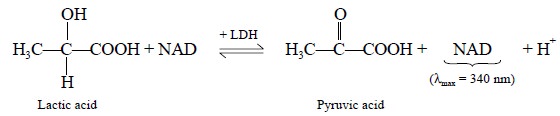
The liberated
nicotinamide-adenine-dinucleotide hydrogenase (NADH) has an absorption maxima
at 340 nm, whereas lactic acid. NAD+ and pyruvic acid do not absorb
at all at this wavelenath.
Temperature Correction Factor :
The
rate of the above reaction is temperature dependent. Hence, if the temperature (experimental) is
higher or lower than that used to define a unit of activity, a definite
correction factor should be applied as per Table 2.10.
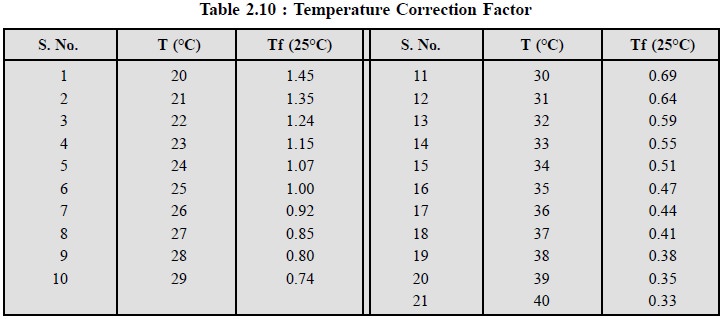
From Table 2.10 it may be observed that :
(a) At a
temperature beyond 25°C (Tf = 1.0), the absorbance increases at a faster rate
than at 25°C due to enhanced rate of reaction and enhanced formation of NADH,
thereby lowering the correction factor from 1.0 e.g., 0.80 at 28°C,
(b) At a
temperature lower than 25°C the rate of reaction is slower than at 25°C,
thereby increasing the correction factor from 1.0 e.g., 1.24 at 24°C, and
(c) Rule of
thumb suggests that for each 10°C rise in temperature the reaction rate is
almost doubled and the correction factor is halved, for example : at 35°C the
correction factor is 0.47 (or 1.0/2 −~ 0.47).
Profile of LDH-levels :
1)
Normal LDH levels are as follows : Absorbance Units per
ml : 42 to 130, International Units per ml : 0.20 to 0.063
2)
LDH level in serum is found to be increased in 8 to 10
hours after a myocardial infarction (i.e.,
development or presence of an infarct in the heart) ; obviously the heart
muscle is destroyed and consequently the enzymes leak into the serum,
3)
Increased LDH levels are found in patients suffering from
diseases related to liver and renal func-tions, cancer and pulmonary
infarction,
4)
Drugs like codeine and morphine help in enhancing LDH
levels.
Materials Required :
Dermatube LDH provided by
Worthington Biochemical, USA.
Procedure :
The following steps need to be
followed in a sequential manner :
1)
Dissolve the contents of Dermatube LDH (containing NADH
and lactic acid) with 2.8 ml of DW,
2)
Put this solution in a cuvette and then insert it in a
colorimeter previously warmed up to 25°C. Set the wavelength at 340 nm.
Carefully adjust the absorbance of this solution to 0.1 by making use of the
proper variable control as explained earlier,
3)
Remove the cuvette and add to it 0.2 ml of serum. Mix the
contents of the cuvette and replace it quickly in position. Carefully record
the absorbance exactly at intervals of 30 seconds for 2 to 3 minutes. In case,
the absorbance happens to rise very rapidly, repeat step 3 by diluting 0.1 ml
of the serum to 0.2 ml with DW,
4)
From the foregoing measurement of absorbances calculate
an average ∆A/min,
5)
Note the temperature at which the reaction is carried out
accurately and then find out Tf from Table 2.10.
6)
Report the LDH concentration as follows :
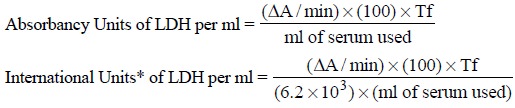
Related Topics