Chapter: Biochemical Pharmacology : Pharmacology of nitric oxide (NO)
Biochemical mechanisms of NO signaling
Biochemical mechanisms of NO
signaling
How does NO signalling work?
As mentioned above, NO is generated within the cytosol of the endothelial cell
(or, in the CNS, the presynaptic cell). As it is able to cross cell membranes
with ease, it will diffuse into neighbour-ing cells, i.e the smooth muscle
cells (in the blood ves-sel walls) or the post-synaptic nerve cells (in the
case of nNOS). There, it will bind to a heme group that is attached to the
enzyme called `soluble guanylate cyclase' (sGC). NO binding will activate sGC,
which will result in the syn-thesis of cyclic guanosine monophosphate (cGMP)
from GTP, in a manner analogous to adenylate cyclase, which as we've seen forms
cAMP from ATP. However, the molec-ular mechanism of sGC activation by NO is
quite special: Binding of NO to one side of the heme moiety of sGC will break
the bond of the heme iron to a histidine residue on the opposite side, which in
turn triggers the conformation-al change that leads to activation (Figure
11.5a). The heme moiety seems to be there solely for the purpose of NO bind-ing
but not to be part of the active site.
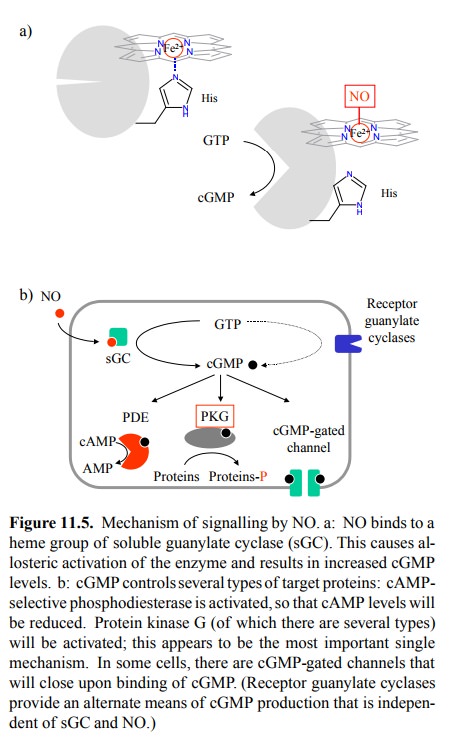
cGMP is a second messenger
with a variety of effector mechanisms (Figure 11.5b), the foremost one of which
is the activation of a cognate protein kinase2, commonly referred to
as `protein kinase G' (PKG; G for cGMP-dependent). In addition, cGMP also
controls ligand-gated ion channels in sensory cells as well as in nerve cells
and smooth muscle cells; not much is know on the contribution of such channels
to NO signalling. Finally, cGMP activates phosphodiesterase, which will
inactivate cAMP by cleav-age to AMP. Thus, cGMP is somewhat of an antagonist of
cAMP.
How does cGMP bring about
relaxation of vascular smooth muscle? Several mechanisms, all of which involve
PKG, have been proposed; none of them is at present certain to be `the' major
one. Moreover, while several relevant changes in protein phosphorylation have
been observed, it is not clear at present whether they are caused by PKG
directly or by intervening secondary kinases.
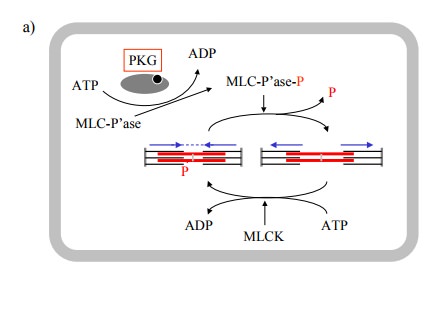
In smooth muscle, contraction
is controlled by the phospho-rylation state of the myosin regulatory light
chain. The ex-tent of this phosphorylation will depend on the regulatory states
of both myosin light chain kinase (MLCK) and ofmyosin light chain phosphatase.
It seems that PKG phos-phorylates the phosphatase, thereby increasing its
activity (Figure 11.6a).
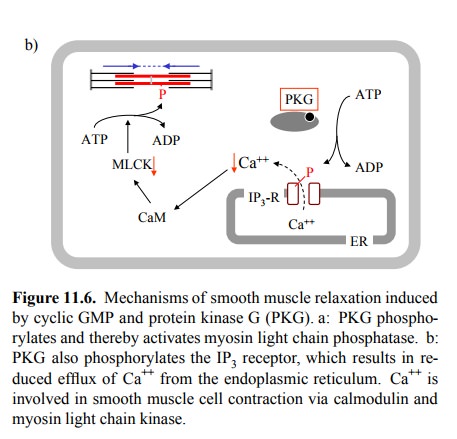
Another
plausible candidate effector molecule for cGMP / PKG is the IP3
receptor channel in the endoplasmic reticu-lum (Figure 11.6b). Its
phosphorylation reduces release of Ca++ from the ER in response to,
e.g., adrenergic stimuli (recall that α1-receptors signal through IP3 / Ca++). The
two mechanisms combined would cause both decreased phos-phorylation and
increased dephosphorylation of myosin light chains. There is also experimental
evidence indicat-ing that phospholipase C is inhibited by PKG, which would lead
to inhibited formation of IP3, and that Ca++-ATP'ases in
both the ER and the cytoplasmic membrane may be acti-vated. Since Ca++-ATP'ases
remove Ca++ from the cytosol, this would further reduce the
availability of Ca++ for con-traction. While all these mechanisms
appear plausible, it is impossible at present to assess their relative
importance in the effect of cGMP- and NO-mediated signalling. Howev-er, given
the very strong relaxing effect of NO on the vas cular smooth muscle, it is reasonable
to assume that more than one mechanism contributes significantly.
The second aspect of NO that
needs to be considered is its chemical reactivity. In fact, NO binds similarly
well to the heme moieties in hemoglobin and in guanylate cyclase, and reaction
with hemoglobin is often considered a major mechanism of inactivation of NO.
However, NO bound to non-oxygenated heme may be released again, so that
hemoglobin may actually serve as a carrier. Alternative-ly, NO may be
transferred from heme to protein cysteine sulfhydryl groups. One quite reactive
such cysteine residue is located within hemoglobin itself. By removing NO from
heme, the cysteine becomes S-nitrosylated itself.
Heme does, however, not seem
to be required for protein S-nitrosylation to occur. The precise chemistry is
unsettled; Figure 11.7a gives one hypothetic reaction scheme. Here,
S-nitrosylation generates one equivalent of superoxide, which in turn (and
particularly so at high concentrations of NO) may react with another molecule
of NO to generate peroxynitrite. Peroxynitrite is very reactive and may
oxi-dize other sites in proteins, or it may give rise to O-nitrosy-lation of
protein tyrosine side chains.

While different reaction
mechanisms for S-nitrosylation have been proposed, a common motif in all of
those I have seen is the participation of oxygen, which makes sense, as the
hydrogen of the sulfhydryl group must be disposed of. NO groups can also be
transferred from one sulfhydryl group to another (Figure 11.7b), so that
covalently bound NO is rarely ever excluded from further circulation. This also
means that protein S-nitrosylation should be reversible by way of transferring
the nitrosyl group to a low-molec-ular weight thiol compound such as, e.g.,
free cysteine or glutathione.
That
S-nitrosylation indeed occurs in vivo is illustrated in Figure 11.8. In the
experiment depicted, the S-nitrosylated proteins have been visualized using an
antibody that specifi cally recognizes the -S-N=O group as its epitope. This
anti-body is targeted with a secondary antibody, which in turn is coupled to an
enzyme. The latter releases a coloured, insol-uble product from a soluble
`chromogenic' precursor (Fig-ure 11.8a). Localized precipitation of the
insoluble stain will thus highlight the distribution and extent of protein
S-nitrosylation.
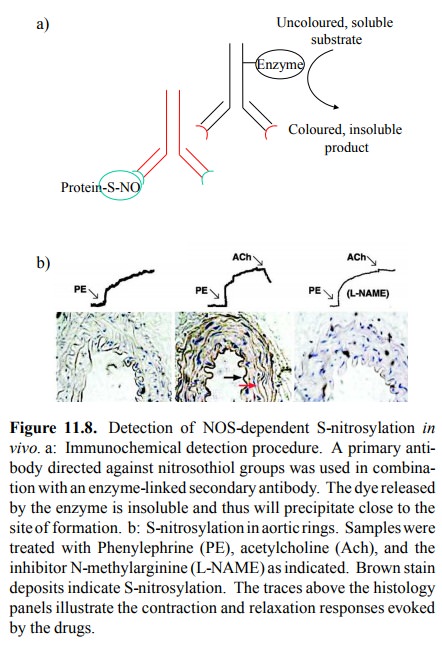
As expected, the most
intensely stained region is the en-dothelium itself (Figure 11.8b, middle
panel); this makes sense, as the concentration of NO should be highest at the
site of formation. However, the surrounding smooth mus-cle tissue is stained as
well, suggesting that indeed S-nitro-sylation might contribute to the muscle
relaxation triggered by NO. That nitrosylation is indeed due to the activity of
NOS, and that detection is reliable is evident from the fact that in the
presence of an inhibitor of NOS no stain is accu-mulated (Figure 11.8b, right
panel).

Does
S-nitrosylation really have a regulatory role in the cell? This raises the
question how specific protein S-ni-trosylation might be. From the non-enzymatic
nature of the chemistry discussed above, one might expect S-nitro-sylation to
be a very indiscriminate process, which would amount to a lot of `noise' in the
signal – and thus, probably, no real signal at all, just noise. The number and
identity of proteins affected by S-nitrosylation has been studied using a
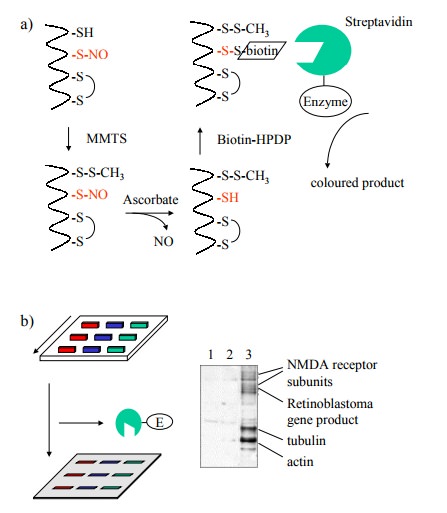
rather ingenious experimental approach, outlined in Fig-ures 11.9 and 11.10. In
these experiments, the nitrosylated sulfhydryl groups were derivatized with
biotin, which fa-cilitates selective detection and / or purification 3.
The chal-lenge here consists in avoiding those cysteine residues that are
either free or part of disulfide bonds. The free ones were first blocked with
the reagent methylmethanethiol-sulfonate (MMTS). While one way to reduce nitrosothiol
groups would be reaction with an excess of low molecular weight thiol (such as
dithiothreitol or 2-mercaptoethanol), this would also reduce the disulfide
bonds (both the cystines and the newly formed MMTS derivatives). Selective
reduc-tion of nitrosothiols only can be achieved using ascorbic acid; in this
way, only the formerly nitrosylated cysteines will be amenable to
biotinylation. Biotinylated proteins can be selectively detected, after gel
electrophoresis and blot-ting, with strepavidin that is coupled to an enzyme,
again using a chromogenic substrate (Figure 11.9).
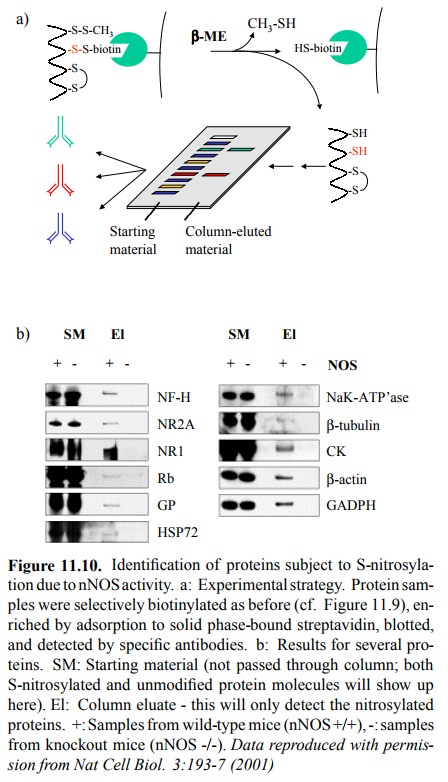
Figure 11.9b shows that a
spectrum of proteins is affect-ed by S-nitrosylation within a sample from
neuronal cells. The nitrosylating agent, in this case, was not NO itself but S-nitroso-glutathione,
illustrating the fact that indeed the NO moiety may travel easily between
sulfhydryl groups (cf Figure 11.7b). Given the fact that most proteins should
have one or more free cysteine residues, the number of proteins that are
detectably labeled is surprisingly small. Stained bands were recovered from the
blots and identified by `pro-teomics' methods, i.e. proteolytic fragmentation,
mass spectrometry, and Edman degradation. Some of the names of identified
proteins seem to `ring a bell' with respect to possible involvement in signal
transduction cascades – e.g., ion channels (including the NMDA subtype
glutamate re-ceptor); others, however, don't (e.g. tubulin, actin4).
The same
authors also examined the S-nitrosylation of pro-teins by nNOS, in neuronal
tissue from mice (Figure 11.10). Conversion of nitrosylated to biotinylated
cysteines was done as above. Biotinylation was, in this case, used to ex-tract
the (formerly) nitrosylated proteins from the total mix-ture of cellular
proteins by selective binding to solid-phase attached streptavidin. After
retrieving the bound material by reduction with excess free thiol, the samples
were run on a gel, blotted, and individual proteins detected using an tibodies
directed not against S-N=O (it's no longer there, is it) but against those
proteins themselves. Figure 11.10b shows a roundup of several nitrosylated
proteins. For com-parison, the starting material (the total protein extract,
with-out pre-selection by streptavidin binding) was run next to the samples
retrieved from the streptavidin column. Fur-thermore, dependency of
nitrosylation on NOS activity was confirmed by parallel processing of samples
from nNOS knockout mice (nNOS -/-).
What does all this tell us?
It has
been claimed that S-nitrosylation may have a similar-ly fundamental regulatory
role as protein phosphorylation has. Is this claim substantiated by the
findings presented? My personal impression is that it is not. However, this
area is presently under intense investigation, and protein nitro sylation is
considered by many as a viable mechanism of signal transduction.
Related Topics