Chapter: Pharmaceutical Biotechnology: Fundamentals and Applications : Monoclonal Antibodies in Cancer
Bevacizumab - Classes of Monoclonal Antibodies: Vascular Endothelial Growth Factor Inhibitors
CLASSES OF MONOCLONAL ANTIBODIES: VASCULAR ENDOTHELIAL GROWTH FACTOR INHIBITORS
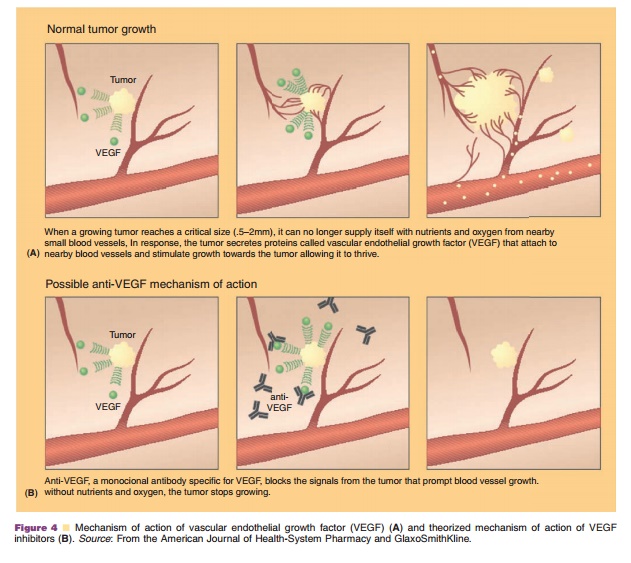
Angiogenesis inhibitors have been in clinical devel-opment for decades, based on the theory by Folkman that the development of new blood vessels (i.e., angiogenesis) must occur for tumor growth beyond 1–2 mm3(Folkman, 1971, 1995; Miller, 2002). As tu-mors enlarge, the centers become hypoxic and stimulate angiogenic growth factors, as shown in Figure 4. VEGF is thought to be one of the most potent growth factors and has been shown to induce neovascularization for malignant cells in an autocrine fashion (Gordon et al., 2001). High levels of VEGF have also been correlated with poor prognosis, disease recurrence, and metastases in a variety of neoplasms.
Bevacizumab
Pharmacology and Mechanism of Action
Bevacizumab (Avastin ) is currently the only FDA approved VEGF inhibitor available. It is a huma-nized MAb that consists of a normal human IgG1(93%) and the VEGF binding residues from a murine neutralizing antibody (7%) (Gordon et al., 2001; Yang et al., 2003). Bevacizumab is produced in a system containing Chinese hamster ovary mammalian cell expression and a gentamicin-containing nutrient medium (Genentech, 2006a).
In vivo and in vitro studies have shown that bevacizumab inhibits the biologic activities of VEGF and does not allow any VEGF isoforms to bind to its receptors (Margolin et al., 2001). This halts tumor growth, as shown in Figure 4. Preclinical trials in animal models have demonstrated the suppression and stabilization of multiple tumor lines through bevacizumab administration.
Bevacizumab has demonstrated both cytostatic and cytotoxic effects in clinical trials (Cobleigh et al., 2003). Specifically, objective responses, a reduction in tumor growth, an increase in time to tumor progres-sion, and an increase in overall survival have been documented in various solid tumor states (Yang et al., 2002, 2003; Kabbinavar et al., 2003).
Pharmacokinetics/Pharmacodynamics
After a single dose of bevacizumab (0.1–10 mg/kg), the maximum concentration (Cmax) ranges from 2.80 to 284 mg/mL, showing a dose-response increase (Gordon et al., 2001). AUC values range from 31 to 87 mg/mL for a 0.3 mg/kg dose up to 2,480–6,010 mg/mLfor a 10 mg/kg dose (Gordon et al., 2001). Multidosing studies have demonstrated an accumulation ratio of 2.8 when 10 mg/kg of bevacizumab is administered every 14 days (Gordon et al., 2001). Steady state concentrations are reached in approximately 100 days (Genentech, 2006a).
Bevacizumab demonstrates linear kinetics over a dosing range of 3–10 mg/kg, a limited volume of distribution, and a low clearance (Gordon et al., 2001). The volume of distribution is 45.7 mL/kg or 3260 mL (Gaudreault, 2001) with a serum half-life of approxi-mately 13–21 days (Gordon et al., 2001; Margolin et al., 2001). Clearance appears to vary based on gender, tumor burden, body weight, and albumin levels (Gaudreault, 2001; Genentech, 2006a). Males have a higher clearance (CL) and larger volume of distribution (Vd) compared to females (CL: 0.262 L/day vs. 0.207 L/ day, Vd: 3.25 L vs. 2.66 L, respectively) after body weight adjustments (Genentech, 2006a). Patients with higher tumor burdens have a higher clearance com-pared to those with lower burdens (CL: 0.249 L/day vs. 0.199 L/day) (Genentech, 2006a). Additionally, a 30% change in body weight results in an 18% change in clearance (Hsei et al., 2002). Finally, reduced albumin levels increase bevacizumab clearance (Gaudreault, 2001). Despite the differences seen in gender, tumor burden, weight and albumin levels, no significant differences are seen in response rate or toxic effects and no dosing modifications are needed.
Many malignant cell lines have been shown to produce VEGF (Gordon et al., 2001). High levels of free VEGF correlate with a poor prognosis and an increased risk of metastatic disease. Most laboratory assays measure total and free VEGF serum concentra-tions. An increase in the concentration of total VEGF serum concentration is observed during bevacizumab treatment (Gordon et al., 2001; Margolin et al., 2001; Yang et al., 2003). This may be due to decreased clearance of VEGF-bound inactive bevacizumab, as this laboratory assessment does not distinguish between free and bound VEGF. At single doses greater than 0.3 mg/kg, phase I studies demonstrated complete suppression of free serum VEGF that remained undetectable throughout the study (Gordon et al., 2001; Margolin et al., 2001). More recent investigations suggest that plasma VEGF levels are more accurate to assess of circulating VEGF, but are not as widely available (Hsei et al., 2002).
Currently, there is no clear association between the dose of bevacizumab, disease stability, or toxicity. However, a phase I trial did note that bevacizumab patients with stable disease had slightly elevated baseline VEGF levels compared to bevacizumab patients demonstrating progressive disease (Gordon et al., 2001). The authors hypothesized that elevated baseline VEGF levels may indicate patients that may be better candidates for bevacizumab therapy.
Indications and Clinical Efficacy
Bevacizumab is currently approved by the FDA for the first or second-line treatment of patients with metastatic carcinoma of the colon and rectum (incombination with intravenous 5-fluorouracil-based chemotherapy) and for the first-line treatment of locally advanced, recurrent, or metastatic nonsmall cell lung cancer (NSCLC) in combination with carboplatin and paclitaxel (Genentech, 2006a).
Bevacizumab, as an additive to standard che-motherapy, appears to significantly prolong overall survival in metastatic colorectal patients. Two multi-centered, randomized phase III trials provided the bulk of efficacy and safety evidence leading to FDA’s approval of bevacizumab as part of first- and second-line chemotherapy in metastatic colorectal cancer (CRC) patients. The first trial randomized 925 pre-viously untreated patients to one of three arms: (i) irinotecan, 5 fluorouracil (5-FU), leucovorin (LV) (Saltz regimen), (ii) Saltz plus bevacizumab, or (iii) 5-FU, LV
plus bevacizumab (Hurwitz et al., 2003). The Saltz plus bevacizumab arm demonstrated statistically signifi-cant advantages compared to the Saltz regimen alone in overall survival (20.3 vs. 15.6 months), progression-free survival (10.6 vs. 6.24 months), OR (45% vs. 35%), and duration of response (10.4 vs. 7.1 months) (Dearden et al., 2001). Grade 3/4 toxicities were similar between each arm. The second phase III trial, E3200, investigated oxaliplatin, 5-FU, LV (FOLFOX4) versus FOLFOX4 plus bevacizumab versus bevacizumab alone in 757 pre-treated metastatic colorectal patients (Benson et al., 2003). Overall survival, time to progression, and OR all significantly favored bevacizumab plus FOLFOX4 vs. FOLFOX alone. Bevacizumab monotherapy was not effective in this patient populationBevacizumab has also shown promising results with combination chemotherapy in patients with advanced NSCLC. The addition of bevacizumab to paclitaxel plus carboplatin in the first-line treatment of advanced NSCLC significantly prolonged overall survival by > 2 months in over 878 patients rando-mized to receive triplet therapy versus the paclitaxel and carboplatin combination (12.3 vs. 10.3 months, respectively) (Genentech, 2006a). However, it must be noted that patients with central tumors with a squamous histology were excluded as phase II data showed that these patients were more likely to develop hemoptysis (Devore et al., 2000; Kabbinavar et al., 2001; Novotny et al., 2001). Post hoc analyses did show that bevacizumab therapy was less effective in women, patients older than 65 years of age, and those with greater than 5% weight loss from the study’s start (Genentech, 2006a).
Since angiogenesis is an important step in the proliferation of breast cancer cells (Cobleigh et al., 2003), bevacizumab has been investigated in two large scale phase III trials in women with breast cancer (Miller et al., 2002; Hillan et al., 2003; Miller, 2003). The first trial compared capecitabine monotherapy to capecitabine plus bevacizumab in 462 metastatic breast cancer patients. No statistically significant difference was found for the primary endpoint; time to disease progression (4.17 months for the control vs. 4.86 months for bevacizumab arm; hazards ratio: 0.98). However, the OR rate, a secondary endpoint, was favorable for the bevacizumab arm (19.8% vs. 9.1%). The second phase III trial is still ongoing and is investigating bevacizumab in locally recurrent breast cancer patients (Miller, 2003), as it is theorized that antiangiogenic therapy in less advanced patients may halt the spread of metastatic disease. A total of 722 locally recurrent or metastatic patients were randomized to receive paclitaxel alone or in combina-tion with bevacizumab. Interim results show that a significant 5-month extension in time to progression is offered by the combination (11 months) compared topaclitaxel alone (6 months) (Lyseng-Williamson and Robinson, 2006).
Safety Concerns
The most common adverse events in all clinical trials have been headache, nausea, vomiting, anorexia, stomatitis, constipation, upper respiratory infection, epistaxis, dyspnea, and proteinuria (Genentech, 2006a). No patients have developed antibodies to bevacizumab during clinical trials (Gordon et al., 2001).
The most serious adverse events associated with bevacizumab treatment include hypertensive crises, nephrotic syndrome, hemorrhage, gastrointestinal perforations, wound healing complications, and con-gestive heart failure (Genentech, 2006a).
The development of hypertension is hypothe-sized to occur through a decreased production of nitric oxide (NO) through VEGF receptor blockade (Cobleigh et al., 2003). The manufacturer recommends that patients treated with bevacizumab should have their blood pressure monitored every 2 to 3 weeks during treatment. During clinical trials, treatment of bevacizumab-associated hypertension included diurectics, calcium channel blockers, beta blockers, and angiotensin-converting enzyme inhibitors. Bevacizumab should be discontinued if patients develop hypertensive crises (Genentech, 2006a).
Proteinuria was an early concern in phase I and II trials, although it has not been associated with renal impairment. The manufacturer recommends monitor-ing for the development or worsening of proteinuria through serial urinalyses (Genentech, 2006a). Patients with a urine dipstick reading of 2 þor greater should be further evaluated. Nephrotic syndrome has been reported in 5/1032 (0.5%) bevacizumab patients (Genentech, 2006a). Bevacizumab should be discon-tinued in patients with nephrotic syndrome.
One area of concern with antiangiogenesis inhibitors is vascular dysfunction, as VEGF regulates vascular proliferation and permeability (Kilickap et al., 2003). The package insert contains a boxed warning regarding the risk of wound healing compli-cations, hemorrhage, and gastrointestinal perforations (Genentech, 2006a). Bevacizumab has shown varying adverse effects on the vasculature including bleeding, thrombotic events, wound healing complications, and gastrointestinal perforations. Bevacizumab therapy should not be initiated within 28 days of major surgery and should be stopped prior to elective surgery. It is unknown how soon before elective surgery that therapy should be halted, but the manufacturer recommends that clinicians take into consideration the long half-life (i.e., 20 days) of bevacizumab (Genentech, 2006a).
A phase II trial comparing carboplatin, paclitaxel (CP) plus bevacizumab to CP alone in 99 Stage IIIb/IV
NSCLC patients uncovered life-threatening hemopty-sis in 6 patients receiving bevacizumab (DeVore et al., 2000). This was fatal in four patients. A subset analysis identified squamous cell histology and central, cavitary, or necrotic tumors as risk factors (Novotny et al., 2001). To prevent further cases, patients with a previous history of hemoptysis should not receive bevacizumab (Gray et al., 2003; Sparano et al., 2004).
Pharmaceutical Considerations: Formulation, Other Routes of Administration, Dosing Regimens
Bevacizumab should be administered as an IV infusion given after the completion of chemotherapy (Genentech, 2006a). After the infusion, the line should be flushed with 0.9% sodium chloride equal to the amount present in the intravenous tubing.
The initial dose should be given over 90 min. No premedications are required. If this is tolerated, the next infusion may be given over 60 min. Subsequent infusions may then be infused over 30 min. Less than 3% of patients had infusion reactions with initial bevacizumab regimens (Genentech, 2006a). Some institutions are now administering bevacizumab in a shorter amount of time based on lack of infusion reactions. Specifically, Memorial Sloan Kettering Cancer Center administers a 5 mg/kg dosage over 10 min, a 10 mg/kg dose over 20 min, and 15 mg/kg over 30 min (Saltz et al., 2006).
Preparation of bevacizumab includes withdraw-ing the necessary amount of bevacizumab for a 5 mg/ kg dose and diluting it with 0.9% sodium chloride for injection for a total volume of 100 mL (Genentech, 2006a). Vials should be discarded within 8 hr after entry. Dextrose solutions should never be used as diluents or administered concomitantly. Both poly-ethylene and polyvinyl bags may be used for administration.
The FDA approved dosages are 5–10 mg/kg administered every 14 days for CRC and 15 mg/kg every 3 weeks for NSCLC (Genentech, 2006a).
Related Topics