Chapter: Modern Pharmacology with Clinical Applications: Antineoplastic Agents
Antimetabolites: Pyrimidine Analogues
Pyrimidine Analogues
Cytarabine
Cytarabine (cytosine
arabinoside, ara-C, Cytosar-U) is an
analogue of the pyrimidine nucleosides cytidine and deoxycytidine. It is one of
the most active agents avail-able for the treatment of acute myelogenous
leukemia. Cytarabine kills cells in the
S-phase of the cycle by com-petitively inhibiting DNA polymerase. The drug
must
first be activated by
pyrimidine nucleoside kinases to the triphosphate nucleotide ara-cytosine
triphosphate (ara-CTP). The susceptibility of tumor cells to cytara-bine is
thought to be a reflection of their ability to acti-vate the drug more rapidly
(by kinases) than to inacti-vate it (by deaminases).
Cytarabine is rapidly
metabolized in the liver, kid-ney, intestinal mucosa, and red blood cells and
has a half-life in plasma of only 10 minutes after intravenous bolus injection.
The major metabolite, uracil arabi-noside (ara-U), can be detected in the blood
shortly af-ter cytarabine administration. About 80% of a given dose is excreted
in the urine within 24 hours, with less than 10% appearing as cytarabine; the
remainder is ara-U. When the drug is given by continuous infusion, cytarabine
levels in CSF approach 40% of those in plasma.
Cytarabine is used in the
chemotherapy of acute myelogenous leukemia, usually in combination with an
anthracycline agent, thioguanine, or both. It is less use-ful in acute
lymphoblastic leukemia and the lymphomas and has no known activity against
other tumors. It has been used intrathecally in the treatment of meningeal
leukemias and lymphomas as an alternative to meth-otrexate.
Myelosuppression is a major
toxicity, as is severe bone marrow hypoplasia. Nausea and mucositis also may
occur. Intrathecal administration occasionally pro-duces arachnoiditis or more
severe neurological toxicity.
Fluorouracil
Fluorouracil (5-fluorouracil,
5-fluorouracil, Efudex, Adrucil) is a halogenated pyrimidine
analogue that must be activated
metabolically. The active metabolite that inhibits DNA synthesis is the
deoxyribonucleotide 5-fluoro-2’ deoxyuridine-S -phosphate (FdUMP). 5-Fluorouracil is selectively toxic to
proliferating rather than non-proliferating cells and is active in both the G1-and
S-phases. The target enzyme inhibited by 5-fluo-rouracilfluorouracil is
thymidylate synthetase, which catalyzes the following reaction:
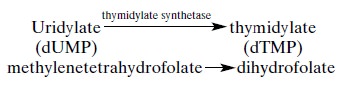
The carbon-donating cofactor
for this reaction is N5,N10 methylenetetrahydrofolate,
which is converted to dihydrofolate. The reduced folate cofactor occupies an
allosteric site on thymidylate synthetase, which al-lows for the covalent
binding of 5-FdUMP to the active site of the enzyme.
Another action proposed for
5-fluorouracil may in-volve the incorporation of the nucleotide
5-fluorouri-dine triphosphate (5-FUTP) into RNA. The cytotoxic role of these
“fraudulent” 5-fluorouracil-containing RNAs is not well understood.
Several possible mechanisms
of resistance to 5-fluo-rouracil have been identified, including increased
syn-thesis of the target enzyme, altered affinity of thymidy-late synthetase
for FdUMP, depletion of enzymes (especially uridine kinase) that activate
5-fluorouracil to nucleotides, an increase in the pool of the normal metabolite
deoxyuridylic acid (dUMP), and an increase in the rate of catabolism of
5-fluorouracil.
The drug has been
administered orally, but absorp-tion by this route is erratic. The plasma
half-life of 5-fluorouracil after intravenous injection is 10 to 20 min-utes.
It readily enters CSF. Less than 20% of the parent compound is excreted into
the urine, the rest being largely metabolized in the liver.
5-Fluorouracil is used in
several combination regi-mens in the treatment of breast cancer. It also has
pal-liative activity in gastrointestinal adenocarcinomas, in-cluding those
originating in the stomach, pancreas, liver, colon, and rectum. Other tumors in
which some antitu-mor effects have been reported include carcinomas of the ovary,
cervix, oropharynx, bladder, and prostate. Topical 5-fluorouracil cream has
been useful in the treatment of premalignant keratoses of the skin and
su-perficial basal cell carcinomas, but it should not be used in invasive skin
cancer.
Floxuridine (FUDR) is the nucleoside
of 5-fluo-rouracil that is readily converted into 5-fluorouracil in vivo. It
has similar pharmacological effects but is pre-ferred to 5-fluorouracil for
hepatic arterial infusions be-cause it is more extensively metabolized in the
liver than 5-fluorouracil, with less systemic toxicity.
The toxicities of
5-fluorouracil vary with the sched-ule and mode of administration. Nausea is
usually mild if it occurs at all. Myelosuppression is most severe after
intravenous bolus administration, with leukopenia and thrombocytopenia
appearing 7 to 14 days after an in-jection. Daily injection or continuous
infusion is most likely to produce oral mucositis, pharyngitis, diarrhea, and
alopecia. Skin rashes and nail discoloration have been reported, as have
photosensitivity and increased skin pigmentation on sun exposure. Neurological
toxic-ity is manifested as acute cerebellar ataxia that may oc-cur within a few
days of beginning treatment.
Related Topics