Chapter: Modern Pharmacology with Clinical Applications: Antineoplastic Agents
Antimetabolites: Folate Antagonists
ANTIMETABOLITES
Folate Antagonists
In general, antimetabolites
used in cancer chemother-apy are drugs that are structurally related to naturally occurring compounds, such as vitamins, amino acids, and nucleotides. These drugs can
compete for binding sites on enzymes or can themselves become incorpo-rated
into DNA or RNA and thus interfere with cell growth and proliferation. The
antimetabolites in clinical use include the folic acid analogue methotrexate,
the pyrimidines (fluorouracil and cytarabine), and the purines (thioguanine,
mercaptopurine, fludarabine, pen-tostatin, and cladribine).
Methotrexate
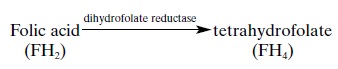
Methotrexate competitively
inhibits the binding of folic acid to the enzyme dihydrofolate reductase.This
enzyme catalyzes the formation of tetrahydrofolate, as follows:
Tetrahydrofolate is in turn
converted to N5,N10-methylenetetrahydrofolate, which is
an essential cofac-tor for the synthesis of thymidylate, purines, methio-nine,
and glycine. The major mechanism by which methotrexate brings about cell death
appears to be in-hibition of DNA synthesis through a blockage of the
biosynthesis of thymidylate and purines.
Cells in S-phase are most sensitive to the cytotoxic ef-fects of
methotrexate. RNA and protein synthesis also
may be inhibited to some extent and may delay pro-gression through the cell
cycle, particularly from G1 to S.
Resistance
Mammalian cells have several
mechanisms of resist-ance to methotrexate. These include an increase in
intracellular dihydrofolate reductase levels, appearance of altered forms of
dihydrofolate reductase with de-creased affinity for methotrexate, and a
decrease in methotrexate transport into cells . The relative importance of each
of these mechanisms of re-sistance in various human tumors is not known.
Cellular uptake of the drug
is by carrier-mediated active transport. Drug resistance due to decreased
transport can be overcome by greatly increasing extra-cellular methotrexate
concentration, which provides a rationale for high-dose methotrexate therapy.
Since bone marrow and gastrointestinal cells do not have im-paired folate
methotrexate transport, these normal cells can be selectively rescued with reduced
folate, bypass-ing the block of dihydrofolate reductase. Leucovorin (citrovorum
factor, folinic acid, 5-formyltetrahydrofo-late) is the agent commonly used for
rescue.
Absorption, Metabolism, and Excretion
Methotrexate is well absorbed
orally and at usual dosages is 50% bound to plasma proteins. The plasma decay
that follows an intravenous injection is triphasic, with a distribution phase,
an initial elimination phase, and a prolonged elimination phase. The last phase
is thought to reflect slow release of methotrexate from tis-sues. The major
routes of drug excretion are glomerular filtration and active renal tubular
secretion.
The formation of polyglutamic
acid conjugates of methotrexate has been observed in tumor cells and in the
liver and may be an important determinant of cyto-toxicity. These methotrexate
polyglutamates are re-tained in the cell and are also potent inhibitors of
dihy-drofolate reductase.
Clinical Uses
Methotrexate is part of
curative combination chemotherapy for acute lymphoblastic leukemias, Burkitt’s
lymphoma, and trophoblastic choriocarci-noma. It is also useful in adjuvant
therapy of breast car-cinoma; in the palliation of metastatic breast, head,
neck, cervical, and lung carcinomas; and in mycosis fungoides.
High-dose methotrexate
administration with leu-covorin rescue has produced remissions in 30% of
pa-tients with metastatic osteogenic sarcoma.
Methotrexate is one of the
few anticancer drugs that can be safely administered intrathecally for the
treat-ment of meningeal metastases. Its routine use as pro-phylactic
intrathecal chemotherapy in acute lym-phoblastic leukemia has greatly reduced
the incidence of recurrences in the CNS and has contributed to the cure rate in
this disease. Daily oral doses of methotrex-ate are used for severe cases of
the nonneoplastic skin disease psoriasis , and methotrexate has been used as an
immunosuppressive agent in severe rheumatoid arthritis.
Adverse Effects
Myelosuppression is the major
dose-limiting toxicity associated with methotrexate therapy. Gastrointestinal
toxicity may appear in the form of ulcerative mucositis and diarrhea. Nausea,
alopecia, and dermatitis are com-mon with high-dose methotrexate. The greatest
danger of high-dose therapy is renal toxicity due to precipita-tion of the drug
in the renal tubules, and the drug should not be used in patients with renal
impairment. Intra-thecal administration may produce neurological toxic-ity
ranging from mild arachnoiditis to severe and pro-gressive myelopathy or
encephalopathy. Chronic low-dose methotrexate therapy, as used for psoriasis,
may result in cirrhosis of the liver. Occasionally meth-otrexate produces an
acute, potentially lethal lung toxi-city that is thought to be allergic or
hypersensitivity pneumonitis. Additionally, methotrexate is a potent te-ratogen
and abortifacient.
Drug Interactions
Salicylates, probenecid, and
sulfonamides inhibit the renal tubular secretion of methotrexate and may
dis-place it from plasma proteins. Asparaginase inhibits protein synthesis and may
protect cells from methotrex-ate cytotoxicity by delaying progression from G1-phase
to S-phase. Methotrexate may either enhance or inhibit the action of
fluorouracil, depending on its sequence of administration.
Gemcitabine
Gemcitabine (Gemzar), an antimetabolite, undergoes
metabolic activation to difluorodeoxycytidine triphos-phate, which interferes
with DNA synthesis and repair. It is the single most active agent for the
treatment of metastatic pancreatic cancer, and it is used as a first-line treatment
for both pancreatic and small cell lung can-cer. It is administered by
intravenous infusion. The dose-limiting toxicity is bone marrow suppression.
Related Topics