Chapter: Clinical Anesthesiology: Anesthetic Management: Respiratory Physiology& Anesthesia
Alveolar, Arterial, & Venous Gas Tensions
ALVEOLAR, ARTERIAL, & VENOUS GAS TENSIONS
When dealing with gas mixtures, each gas
is considered to contribute separately to total gas pressure, and its partial
pressure is directly pro-portional to its concentration. Air has an O2 con-centration of approximately 21%; therefore, if
the barometric pressure is 760 mm Hg (sea level), the partial pressure of O2 (Po2) in air is normally 159.6 mm Hg: 760 mm
Hg × 0.21 = 159.6 mm Hg
In its general form, the equation may be
written as follows:
Pio2= Pb × Fio2
where Pb = barometric pressure and Fio2= the frac-tion of inspired O 2.
Two general rules can also be used:
·
Partial
pressure in millimeters of mercury approximates the percentage × 7
·
Partial
pressure in kilopascals is approximately the same as the percentage.
Oxygen
Alveolar Oxygen Tension
With
every breath, the inspired gas mixture is humidified at 37°C
in the upper airway. The inspired tension of O2 (Pio2) is
therefore reduced by the added water vapor. Water vapor pressure is depen-dent
only upon temperature and is 47 mm Hg at 37°C.
In humidified air, the normal partial pressure of O2 at sea level is
149.7 mm Hg:
(760
−
47) × 0.21 =
149.1 mm Hg
The
general equation is
Pio2=
(Pb − Ph2o) ×
Fio2
where
Ph2o = the vapor pressure of water at
body temperature.
In
alveoli, the inspired gases are mixed with residual alveolar gas from previous
breaths, O2 is taken up, and CO2 is added. The final
alveolar O2 tension (Pao2) is therefore dependent on all
of these factors and can be estimated by the following equation:
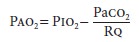
where
Paco2= arterial CO2 tension
and Rq = respiratory quotient.
Rq
is usually not measured. Note that large
increases
in Paco2 (>75
mm Hg) readily pro-duce hypoxia (Pao2<
60 mm Hg) at room air, but not at high inspired O 2 concentrations.
A
yet simpler method of approximating Pao2 in millimeters of mercury
is to multiply the percentage of inspired O 2 concentration by 6.
Thus, at 40%, Pao2 is 6 ×
40, or 240 mm Hg.
Pulmonary End-Capillary Oxygen Tension
For
all practical purposes, pulmonary end-capillary O2 tension (Pc′o2)
may be considered identical to Pao2; the Pao2–Pc′o2
gradient is normally minute. Pc′o2
is dependent on the rate of O2 diffusion across the
alveolar–capillary membrane, as well as on pul-monary capillary blood volume
and transit time. The large capillary surface area in alveoli and the 0.4–0.5 µm
thickness of the alveolar–capillary mem-brane greatly facilitate O2
diffusion. Enhanced O 2 binding to hemoglobin at saturations above
80% also augments O 2 diffusion . Capillary transit time can be
estimated by dividing pulmonary capillary blood volume by cardiac output (pulmo-nary
blood flow); thus, normal capillary transit time is 70 mL ÷
5000 mL/min (0.8 s). Maximum Pc′o2
is usually attained after only 0.3 sec, providing a large safety margin.The
binding of O2 to hemoglobin seems to be the principal rate-limiting
factor in the transferof O2 from alveolar gas to blood. Therefore,
pulmo-nary diffusing capacity reflects not only the capacity and permeability
of the alveolar–capillary mem-brane, but also pulmonary blood flow. Moreover, O2
uptake is normally limited by pulmonary blood flow, not O2 diffusion
across the alveolar–capillary membrane; the latter may become significant
dur-ing exercise in normal individuals at high altitudes and in patients with
extensive destruction of the alveolar–capillary membrane.
O2
transfer across the alveolar–capillary mem-brane is expressed as O2
diffusing capacity (Dlo2):
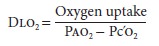
Because
Pc′o2
cannot be measured accurately, measurement of carbon monoxide diffusion
capacity (Dlco) is used instead to assess gas transfer across the
alveolar–capillary membrane. Because carbon monoxide has a very high affinity
for hemoglobin, there is little or no CO in pulmonary capillary blood, so that
even when it is administered at low concen-tration, Pc′co
can be considered zero. Therefore,
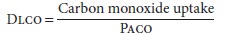
Reductions
in Dlco imply an impediment in gas transfer across the alveolar–capillary
membrane Such impediments may be due to abnormal V/Q
ratios, extensive destruction of the gas alveolar– capillary membrane, or very
short capillary transit times. Abnormalities are accentuated by increases in O2
consumption and cardiac output, such as occurs during exercise.
Arterial Oxygen Tension
Pao2
cannot be calculated like Pao2 but must be measured at room air. T he
alveolar-to-arterial O 2 partial pressure gradient (A–a
gradient) is normally less than 15 mm Hg, but progressively increases with age
up to 20–30 mm Hg. Arterial O 2 tension can be approximated by the
following formula (in mm Hg):
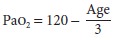
The range is 60–100 mm Hg (8–13 kPa).
Decreases are probably the result of a progressive increase in closing capacity
relative to FRC (see above). Table 23–4 lists the mechanisms of hypox-emia
(Pao2<60
mm Hg).
The most common mechanism for hypoxemia
is an increased alveolar–arterial gradient. The A–agradient for Odepends on the amount of right to-left shunting, the
amount of V/Q scatter, and the mixed venous O2 tension . The last depends on cardiac output, O2 consumption, and hemoglobin concentration.

The A–a gradient for O2 is directly proportional to shunt, but inversely
proportional to mixed venous O2 tension. The effect of each variable on
Pao2 (and consequently the A–a gradient) can
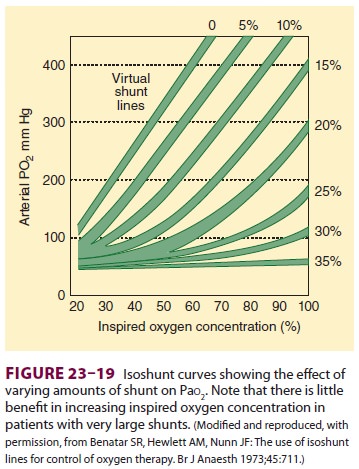
be determined only when the other variables are held constant. Figure 23–19 shows
the effect of different degrees of shunting on Pao2. It should also
be noted that the greater the shunt, the less likely the possibility that an
increase in Fio2 will prevent hypoxemia. Moreover,
isoshunt lines seem to be most useful for O2
concentrations between 35% and 100%. Lower O2
concentrations require modification of isoshunt lines to account for the effect
of V/Q scatter.
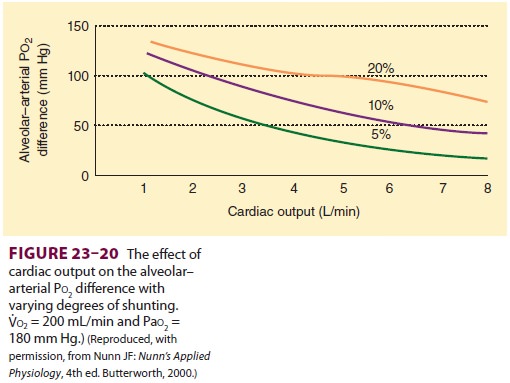
The effect of cardiac output on the A–a
gradi-ent (Figure
23–20) is due not only to its second-ary effects on mixed venous O2 tension but also to a direct relationship between
cardiac output and intrapulmonary shunting. As can be seen, a low car-diac
output tends to accentuate the effect of shunt on Pao2. A reduction in venous admixture may be observed
with low-normal cardiac outputs second-ary to accentuated pulmonary
vasoconstriction from a lower mixed venous O2
tension. On the other hand, high cardiac outputs can increase venous admixture
by elevating mixed venous O2 tension, which in turn inhibits hypoxic
pulmonary vasoconstriction.
O2
consumption and hemoglobin concentra-tion can also affect Pao2 through their secondary effects on mixed venous O2 tension (below). High O2
consumption rates and low hemoglobin
concentra-tions can increase the A–a gradient and depress Pao2.
Mixed Venous Oxygen Tension
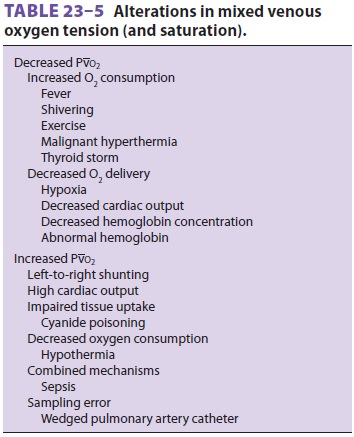
Normal mixed venous O2 tension (Pvo2) is about 40 mm Hg and represents the
overall balance betweenO2consumptionandO![]() delivery (Table 23–5). A true mixed
venous blood sample contains venous drainage from the superior
vena cava, the inferior vena cava, and the heart; it must therefore be obtained
from a pulmonary artery catheter.
delivery (Table 23–5). A true mixed
venous blood sample contains venous drainage from the superior
vena cava, the inferior vena cava, and the heart; it must therefore be obtained
from a pulmonary artery catheter.
2. Carbon Dioxide
Carbon dioxide is a by-product of
aerobic metabo-lism in mitochondria. There are therefore small con-tinuous
gradients for CO2 tension from mitochondria to cell
cytoplasm, extracellular fluid, venous blood, and alveoli, where the CO2 is finally eliminated.
Mixed Venous Carbon Dioxide Tension
Normal mixed venous CO2 tension (Pvco2
) is about 46 mm Hg and is the end result of mixing of blood from tissues of
varying metabolic activity. Venous CO2
tension is lower in tissues with low metabolic activity (eg, skin), but higher
in blood from those with relatively high activity (eg, heart).
Alveolar Carbon Dioxide Tension
Alveolar CO2
tension (Paco2) is generally considered to represent
the balance between total CO production (Vco2)
and alveolar ventilation (elimination):
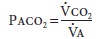
where Va is alveolar ventilation ( Figure 23–21).
In reality, Paco2 is related to CO2 elimination rather than production. Although the
two are equal in a steady state, an imbalance occurs during periods
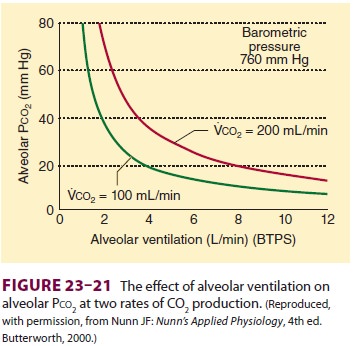
of acute hypoventilation or hypoperfusion, and the excess CO2 increases total body CO2 content.Clinically Pacois moredependentonalveolarventilation than is Vco2, because CO2 output does not vary appreciably under most circumstances. Moreover, the body’s large capacity to store CO buffers acute changes in Vco2.
Pulmonary End-Capillary Carbon Dioxide Tension
Pulmonary end-capillary CO 2 tension (Pc′co2)
is virtually identical to Paco2 for the same reasons as those discussed
in the section about O2. In addition, the diffusion rate for CO2 across the alveolar–capil-lary membrane is 20 times
that of O2.
Arterial Carbon Dioxide Tension
Arterial CO2
tension (Paco2), which is readily mea-surable, is
identical to Pc′co2, and, necessarily, Paco2. Normal Paco2
is 38 ± 4 mm Hg (5.1 ± 0.5 kPa); in practice, 40 mm Hg is
usually considered normal.Although low V/Q
ratios tend to increase•Paco2,whereas high V/Q ratios tend to decrease
it (in contrast to the case for O2 [see above]), significant
arterial-to-alveolar gradients for CO develop only in the presence of marked
V/Q abnormalities (>30% venous admixture); even then the gradient is
rela-tively small (2–3 mm Hg). Moreover, small increasesin the gradient
appreciably increase COoutput into alveoli with relatively normal V/Q. Even
moderate to severe disturbances usually fail to appreciably alter arterial CO 2 because of a reflex increase in ven-tilation from
concomitant hypoxemia.
End-Tidal Carbon Dioxide Tension
Because end-tidal gas is primarily
alveolar gas and Paco2 is virtually identical to Paco2, end-tidal CO 2
tension (Petco2) is used clinically as an estimate of
Paco2. The Paco2–Petco2 gradient is normally less than 5 mm Hg and
represents dilution of alveolar gas with CO2-free
gas from nonperfused alveoli (alveolar dead space).
Related Topics