Chapter: Basic & Clinical Pharmacology : Pharmacokinetics & Pharmacodynamics: Rational Dosing & the Time Course of Drug Action
Bioavailability - Pharmacokinetics

Bioavailability
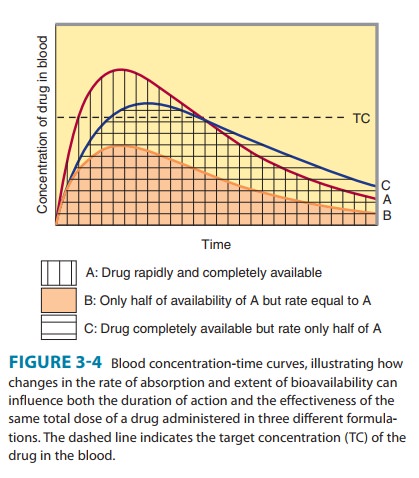
Bioavailability is defined as the fraction of unchanged drug reach-ing the systemic circulation following administration by any route (Table 3–3). The area under the blood concentration-time curve (AUC) is proportional to the extent of bioavailability for a drug if its elimination is first-order (Figure 3–4). For an intravenous dose of the drug, bioavailability is assumed to be equal to unity. For a drug administered orally, bioavailability may be less than 100% for two main reasons—incomplete extent of absorption across the gut wall and first-pass elimination by the liver .
A. Extent of Absorption
After oral administration, a drug may be incompletely absorbed, eg, only 70% of a dose of digoxin reaches the systemic circulation. This is mainly due to lack of absorption from the gut. Other drugs are either too hydrophilic (eg, atenolol) or too lipophilic (eg, acy clovir) to be absorbed easily, and their low bioavailability is also due to incomplete absorption. If too hydrophilic, the drug cannot cross the lipid cell membrane; if too lipophilic, the drug is not soluble enough to cross the water layer adjacent to the cell. Drugs may not be absorbed because of a reverse transporter associated with P-glycoprotein. This process actively pumps drug out of gut wall cells back into the gut lumen. Inhibition of P-glycoprotein and gut wall metabolism, eg, by grapefruit juice, may be associated with substantially increased drug absorption.
B. First-Pass Elimination
Following absorption across the gut wall, the portal blood delivers the drug to the liver prior to entry into the systemic circulation. A drug can be metabolized in the gut wall (eg, by the CYP3A4 enzyme system) or even in the portal blood, but most commonly it is the liver that is responsible for metabolism before the drug reaches the systemic circulation. In addition, the liver can excrete the drug into the bile. Any of these sites can contribute to this reduction in bioavailability, and the overall process is known as first-pass elimination. The effect of first-pass hepatic elimination on bioavailability is expressed as the extraction ratio (ER):

where Q is hepatic blood flow, normally about 90 L/h in a person weighing 70 kg.The systemic bioavailability of the drug (F) can be predicted from the extent of absorption (f ) and the extraction ratio (ER):

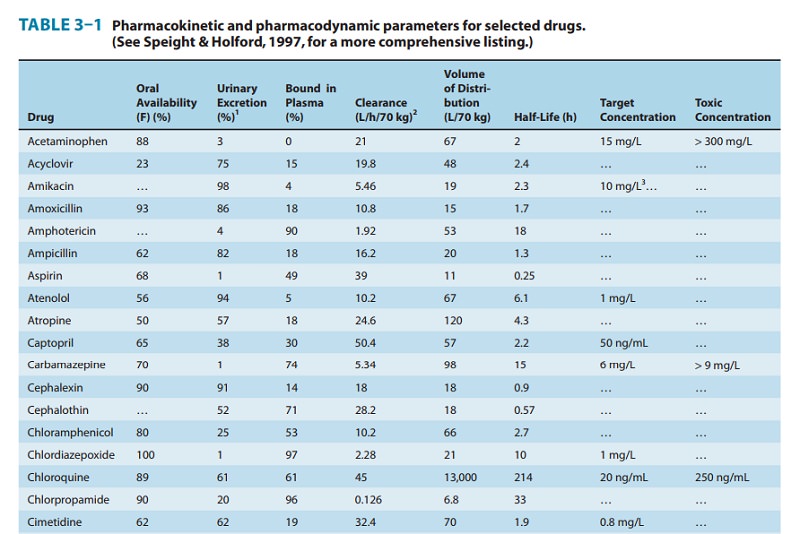
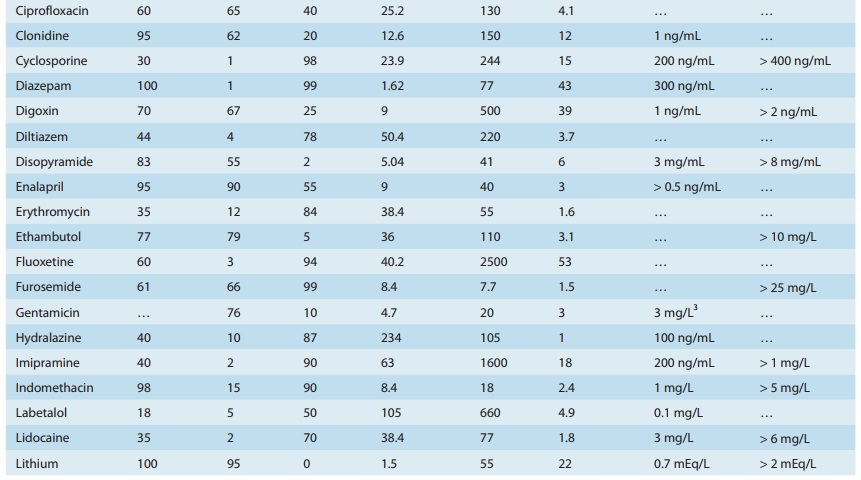
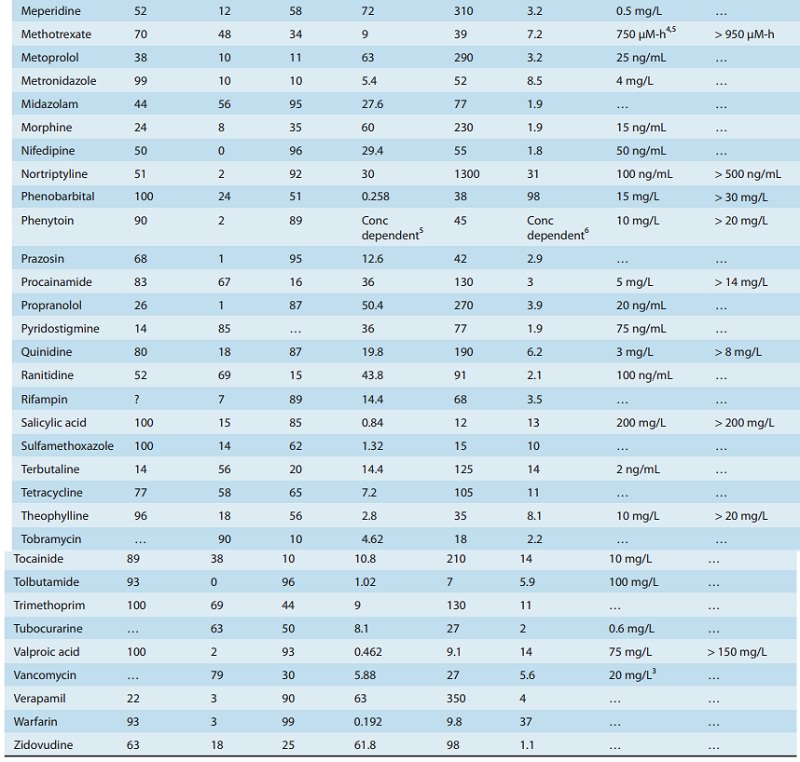
A drug such as morphine is almost completely absorbed (f = 1), so that loss in the gut is negligible. However, the hepatic extrac-tion ratio for morphine is morphine clearance (60 L/h/70 kg) divided by hepatic blood flow (90 L/h/70 kg) or 0.67, so (1 – ER) is 0.33. The bioavailability of morphine is therefore expected to be about 33%, which is close to the observed value (Table 3–1).
C. Rate of Absorption
The distinction between rate and extent of absorption is shown in Figure 3–4. The rate of absorption is determined by the site of administration and the drug formulation. Both the rate of absorption and the extent of input can influence the clinical effectiveness of a drug. For the three different dosage forms depicted in Figure 3–4, there would be significant differences in the inten-sity of clinical effect. Dosage form B would require twice the dose to attain blood concentrations equivalent to those of dosage form A. Differences in rate of absorption may become important for drugs given as a single dose, such as a hypnotic used to induce sleep. In this case, drug from dosage form A would reach its target concentration earlier than drug from dosage form C; concentra-tions from A would also reach a higher level and remain above the target concentration for a longer period. In a multiple dosing regi-men, dosage forms A and C would yield the same average blood level concentrations, although dosage form A would show some-what greater maximum and lower minimum concentrations.
The mechanism of drug absorption is said to be zero-order when the rate is independent of the amount of drug remaining in the gut, eg, when it is determined by the rate of gastric emptying or by a controlled-release drug formulation. In contrast, when the dose is dissolved in gastrointestinal fluids, the rate of absorption is usually proportional to the gastrointestinal concentration and is said to be first-order.
Related Topics