Chapter: Biochemical Pharmacology : Some principles of cancer pharmacotherapy
Antimetabolites
Antimetabolites
Cell
cycle-selective agents comprise antimetabolites (of DNA synthesis) and
inhibitors of mitosis (cell division; see below). A widely used example of an
antimetabolite is 5-fluorouracil (5-FU; Figure 13.11). Before this drug
ac-tually does something interesting, it needs to be converted to the
nucleotide analog 5-fluoro-deoxyuridinemonophos-phate (5-FdUMP), which occurs
in the same way as with normal uracil.

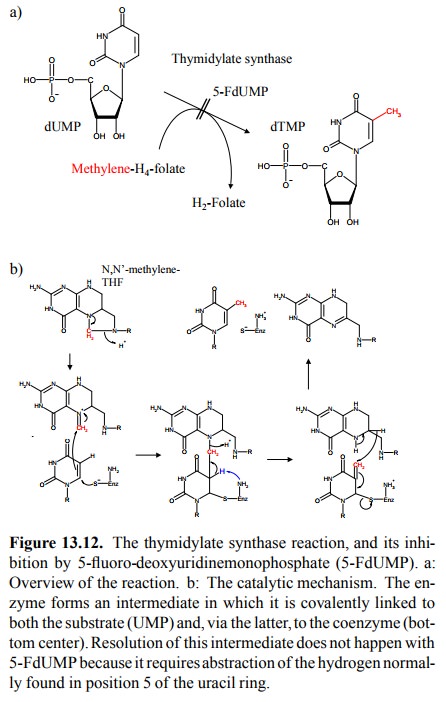
5-FdUMP
is an analog of dUMP, which is the substrate for dTMP synthesis by thymidylate
synthase; it is this reaction that is inhibited by dUMP (Figure 13.12a). The
catalyt-ic mechanism of thymidylate synthase is depicted in Fig-ure 13.12b. The
enzyme (thymidylate synthase) requires N,N'-methylene-tetrahydrofolic acid as a
cosubstrate. The reaction is initiated by a cysteine residue in the active site
of the enzyme and involves an intermediate in which the enzyme, the substrate
(dUMP), and the cosubstrate are all covalently bound (bottom center in Figure
13.12b). This complex is resolved in the second step, which involves
ab-straction of the hydrogen in position 5 of the uracil by a basic residue in
the active site. The trick with 5-FU is that this abstraction doesn't happen,
since position 5 is occupied by fluorine, which is very tightly bound to the
ring. There-fore, the enzyme remains covalently locked up – 5-FU is a covalent
and thus very efficient inhibitor of thymidylate synthase.
Another
intriguing consequence of the fluorine substitution is its promotion of the
tautomeric form of the ring, which has base-pairing properties resembling those
of cytosine. Incorporation of 5-FU (subsequent to further phosphory-lation of
5-FdUMP to 5-FdUTP) therefore induces muta-tions due to misincorporation of guanine
instead of adenine (Figure 13.11c)11. The same behaviour is observed
with the bromine analog of 5-FU (5-bromouracil). Bromine is sim-ilar in size to
a methyl group, so that 5-BU sterically re-sembles thymidine and therefore is
efficiently incorporated into the DNA (more so than 5-FU). It is not used in
cancer therapy but is commonly used (in the form of a pro-drug,
5-bromouracil-deoxyriboside) as a mutagen in experimen-tal research.
We have
just seen that folic acid functions as a coenzyme in the synthesis of dTMP. It
also donates methyl groups in the synthesis of purine bases, so that it is
actually quite impor tant in DNA synthesis. After transfer of the methyl group
by N,N'-methylene-tetrahydrofolate, the remainder (dihy-drofolate) is
regenerated in two steps, the first of which is the reduction to
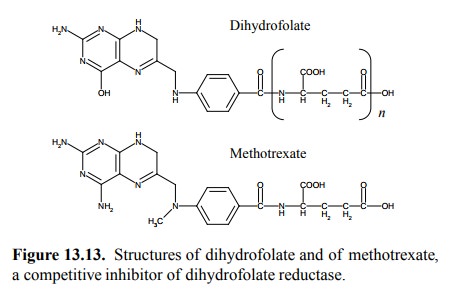
tetrahydrofolate by dihydrofolate reductase. This enzyme is inhibited by
methotrexate (Figure 13.13). Note that with this antimetabolite there is no
possibility of introducing mutagenic base analogs into the DNA. It there-fore
has less carcinogenic potential than most other drugs discussed here and is
also sometimes used as an immuno-suppressive agent in diseases other than
cancer.

A quite unusual
antimetabolite is the enzyme L-asparagi-nase, isolated from E. coli, commonly
used in the treatment of leukemia. Asparagine is a precursor of purine
synthe-sis (Voet & Voet have all the details), and depletion of this amino
acid seems to slow down tumour cells. One could speculate at length why this
would have a preferential ef-fect on tumour cells, but it may be better not. Of
note, this drug can be used for extended periods of time without inducing a
neutralizing immune response (which it nor-mally should) because the immune
system will be quite knocked out under the prevailing conditions of disease and
treatment.
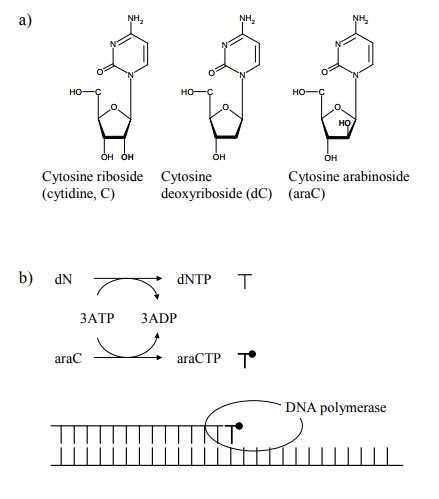
A
nucleotide antimetabolite that carries the modification in the sugar rather
than in the base is cytosine arabinoside (araC; Figure 13.14). In this
molecule, there is an OH group in position 2 of the ribose, pointing in the
`wrong' direction (as compared to ribose). AraC gets incorporated into DNA but
then apparently interferes with further DNA synthesis. This may affect
different DNA polymerases to different extents; in fact, araC reportedly
inhibits DNA repair more strongly than DNA replication (the two processes
involve different DNA polymerases).
Another
aspect of antimetabolite therapy exemplified by araC is the emergence of
resistance in tumours that are ini-tially susceptible. How come? Like 5-FU and
many oth-er antimetabolites, araC requires metabolic activation; on the other
hand, the activated metabolites are also subject to degradation (Figure
13.14c). In the beginning, we noticed that tumour cells are quite instable
genetically. Chromoso-mal deletions or duplications may easily result either in
a re-duced drug activation or in accelerated degradation, by way of changing
the copy numbers of the genes encoding the re-spective enzymes. Similarly,
translocation may cause trans-fer of these genes into foreign regulatory
contexts with con-comitant over- or under-expression. Such causes of cancer
cell drug resistance have been experimentally confirmed.
Related Topics