Chapter: Essential Clinical Immunology: Autoimmunity
Type III Autoimmune Reactions (Immune Complex Disease) - Mechanisms of Autoimmune Tissue Injury
Type III Autoimmune Reactions (Immune Complex Disease)
Autoantibodies also cause disease by form-ing networks of autoantibodies bound to their antigens (immune complexes). The antigen-antibody complexes can deposit in tissues, causing inflammatory lesions. Studies of serum sickness led to the first description of an immune complex dis-ease. Serum sickness is manifested by fever, glomerulonephritis, vasculitis, urti-caria, and arthritis, appearing seven to twenty-one days after primary immuniza-tion or two to four days after secondary immunization with a foreign protein. Two consequences of immune complex forma-tion are complement fixation and binding to Fc or complement receptors on phago-cytes. Clearance is facilitated by the bind-ing of immune complexes to C3b recep-tors (CR1) on erythrocytes, which retain the complexes in the circulation until their removal by the reticuloendothelial cells of the spleen or liver.
Immune complex formation is a nor-mal process that removes foreign antigens from the circulation. Removal of immune complexes by phagocytes bearing Fc or complement receptors prevents their deposition at other sites. The efficiency of uptake of immune complexes by either Fc receptors or CR1 is proportional to the number of IgG molecules associated with the complex.
Immune complexes can activate either the classical or the alternative complement pathway. The classical pathway plays a major role in maintaining immune com-plexes in a soluble form, preventing their deposition in tissues. C3b bound to the sol-ubilized immune complexes promotes their clearance by the erythrocyte complement receptor CR1. If the rate of immune com-plex formation exceeds the ability to clear these complexes via Fc receptors and CR1, the immune complexes can deposit within tissues, leading to inflammation. This efficient immune complex transport and removal by Fc and complement receptors can be overwhelmed, however, leading to tissue deposition and immune complex disease. This situation may result from overproduction of immune complexes, blockade of phagocytosis by the reticulo-endothelial system, or complement deple-tion resulting in inefficient solubilization of immune complexes.
SLE is the prototype of human immune complex disease. Tissue damage in lupus is mainly caused by immune complexes containing autoantibodies to soluble anti-gens. These autoantibodies include anti-bodies against RNA-protein complexes (e.g., anti-Sm, RNP, Ro/SS-A, and La/ SS-B antibodies) and DNA-protein com-plexes (e.g., anti-double-stranded DNA, antihistone, antichromatin antibodies). The target antigens are found mainly in the cell nucleus, although in some cases (e.g., antiribosomal antibodies), they may be cytoplasmic. Immune complexes con-taining these autoantibodies, especially anti-dsDNA antibodies, are selectively enriched in the renal glomeruli (capillary tufts that produce urine as an ultrafiltrate of blood) of patients with lupus nephritis and are thought to play a critical role in establishing the inflammatory response. Immune complex deposition in the kidney leads to proliferative glomerulonephritis and effacement of the normal glomerular architecture (Figure 6.2). As is the case in serum sickness, active lupus nephritis is frequently associated with hypocomple-mentemia (Figure 6.2). In addition to the kidneys (glomeruli), immunoglobulin and complement deposits are found in the blood vessels (vasculitis), skin (rashes), nervous system, and other locations. Pre-formed immune complexes may become trapped in the glomerular filter, or immune complexes may develop in situ because of the interaction of cationic antigens (e.g., histones) with heparan sulfate glycos-aminoglycan in the glomerular basement membrane. The association of lupus with deficiencies of the early classical comple-ment components, especially C2 and C4, is consistent with the role of complement pathways in solubilizing immune com-plexes (see the section “Pathogenesis of Autoimmune Disease”)
Case 2. Systemic Lupus Erythematosus, a Type III Autoimmune Reaction
A fifteen-year-old girl developed myalgias (muscle pain), painful and swollen joints, and low-grade fevers and was found to have a positive anti-nuclear antibodies (ANA) test. Kidney function was normal. She was given a diagnosis of SLE and treated with hydroxychloroquine (an antimalarial), azathioprine (a nucleoside analog), and
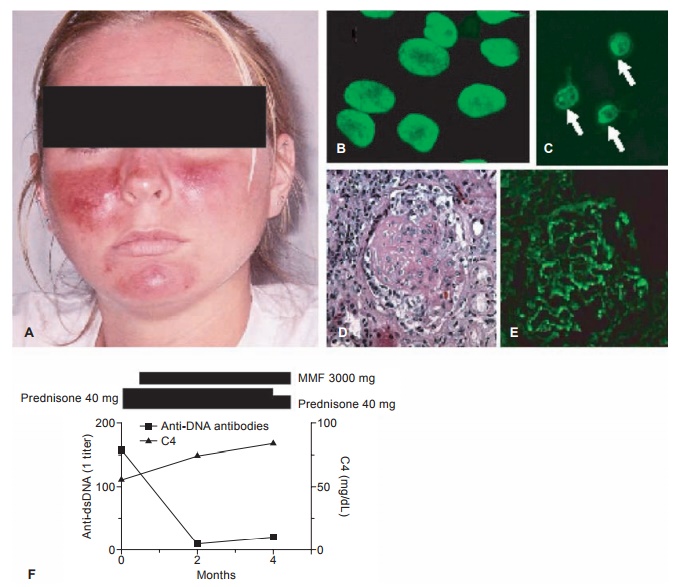
Figure 6.2 Systemic lupus erythematosus. A, Acute cutaneous lupus (malar or butterfly rash); B, fluorescent antinuclear antibody test (HEp-2 cells were stained with patient serum followed by fluorescein isothiocyanate-labeled goat antihuman IgG antibodies); C, Crithidia luciliae kinetoplast staining assay for anti-double-stranded DNA autoantibodies (Crithidia organisms were stained with patient serum followed by fl uorescein isothiocyanate-labeled goat anti-human IgG antibodies). Kinetoplasts are indicated by arrows; D, hematoxylin and eosin staining of a renal glomerulus illustrating proliferative nephritis and crescent formation; E, direct immunofl uorescence of a renal glomerulus in lupus nephritis stained for IgG; F, classic inverse relationship between anti-dsDNA antibody titer (broken line) and complement (C3, solid line) levels. Bars above the graph depict the doses of prednisone and mofetil mycophenolate (MMF) over the same period.
10 mg/day of prednisone (a corticoste-roid). Three years later, she developed alopecia (hair loss) and a red, ulcerat-ing rash of the legs. A skin biopsy was reported to be “consistent with lupus.” The skin lesions resolved when the dose of prednisone was increased. For the next five years, her lupus remained well controlled with hydroxychloro-quine and intermittent low-dose prednisone until she moved to another state and was unable to continue her health insurance. Several months after stop-ping all of her medications, vasculitic skin lesions recurred on the legs, and she developed a rash on the face (Fig-ure 6.2A). Laboratory testing revealed that her creatinine (a measure of renal function) was now abnormally ele-vated at 3.4 mg/dl (normal 1.0 mg/dl), her albumin was low, and her urine tested positive for protein (protein-uria, >300 mg/dl) and blood (hematu-ria). Microscopic examination revealed seven erythrocytes per high-power field. Fluorescent antinuclear antibody testing was positive at a titer of 1:640 homogeneous pattern and anti-dsDNA antibodies were detected at a titer of 1:160 using the Crithidia luciliae kineto-plast staining assay (Figure 6.2B, 6.2C). Complement components C3 and C4 were low (56 and 11 mg/dl, respec-tively). She was treated with a high dose of methylprednisolone (another corticosteroid) intravenously followed by prednisone. A renal biopsy was per-formed and showed proliferative lupus nephritis (Figure 6.2D). Immunofluo-rescence showed staining of the glo-merular basement membrane for IgG (Figure 6.2E) as well as IgM and C3. She was treated with mycophenolate mofetil (MMF, 1,500 mg twice a day), and after four months, her proteinuria and hematuria resolved, the creatinine returned to near baseline (1.1 mg/dL), C3 increased to 85 mg/dl, and anti-dsDNA antibodies decreased to 1:20 (Figure 6.2F).
COMMENT
SLE is the prototype of human immune complex disease. For reasons that are unclear, autoantibodies against dsDNA are involved in the formation of immune complexes that appear to be particularly prone to become trapped in the renal glomeruli, where they can cause inflammation (glomerulo-nephritis). The levels of anti-dsDNA often are low during periods of dis-ease quiescence.
In this case, a flare of disease activity was precipitated by stopping medications that keep the autoimmune response in check (pred-nisone and hydroxychloroquine), leading to the production of high levels of anti-dsDNA antibodies that could be detected by staining the kinetoplast (a circular DNA molecule) of Crithidia luciliae organisms. These autoantibodies formed immune complexes, resulting in the consumption of classical comple-ment components C3 and C4 (the classic inverse relationship between anti-DNA and complement levels, as illustrated in Figure 6.2F). Because the immune com-plexes were inadequately cleared, they deposited in the renal glomeruli, result-ing in the patient’s new onset of hema-turia and proteinuria and the decline in her renal function (increased creati-nine). With reinstitution of appropriate therapy, the anti-DNA levels declined, C3 and C4 levels recovered, and renal immune complex deposition dimin-ished, resulting in an improvement of renal function.
Related Topics