Chapter: Essential Clinical Immunology: Autoimmunity
Pathogenesis of Autoimmune Disease
PATHOGENESIS OF AUTOIMMUNE DISEASE
Genetic Predisposition
Genetic, environmental, and random (sto-chastic) factors all play a role in the patho-genesis of autoimmune diseases. Family members of affected individuals are at higher risk for developing autoimmune disease than the general population. The relative risk to siblings of affected individ-uals (probands) versus the risk in the gen-eral population (λs = disease prevalence in siblings of affected individuals ÷ disease prevalence in the general population) is a useful way to estimate the importance of genetic factors. The relative risk is between five- and fiftyfold higher in siblings of affected probands than in unrelated indi-viduals in most autoimmune diseases (Table 6.3). Part of this effect is accounted for by MHC-linked genes.
Twin studies illustrate the importance of these genetic factors. If the concordance rates in monozygotic and dizygotic twins are about the same, the genetic effect is small. For most autoimmune diseases, concordance rates are 15–30 percent for monozygotic twins versus 2–5 percent for
Table 6.3 Twin Studies IIlustrating the Importance of Genetic Factors in Autoimmune Disease
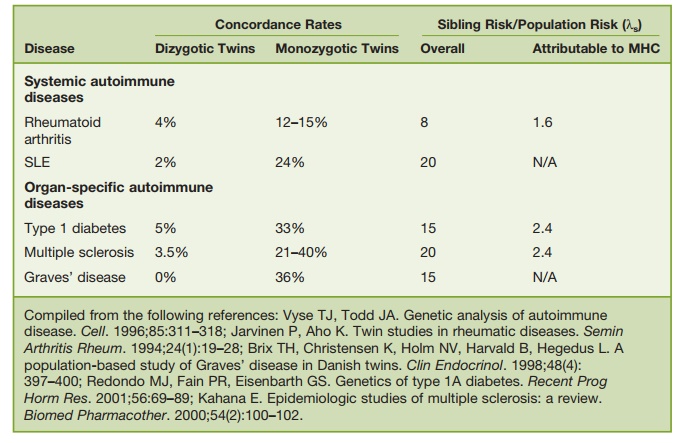
dizygotic twins, consistent with a sizeable genetic effect (Table 6.3). Identification of the actual mutations or genetic polymor-phisms that confer susceptibility to auto-immune diseases has been complicated by the fact that most autoimmune disorders appear to involve multiple genes, each with only a small effect. Moreover, many autoimmune “diseases” are actually “syn-dromes” that may arise through a variety of different pathogenic mechanisms and genetic abnormalities. Even in the inbred lupus-prone mouse strain NZB/W, ten or more susceptibility loci are thought to contribute to disease severity in an addi-tive fashion (threshold liability model). Human SLE and other autoimmune dis-eases also are likely to be highly complex genetically. Interestingly, there may be some overlap genetically between different forms of autoimmune disease, such as SLE and type I diabetes. More than half of the linkages identified in genomewide scan- ning studies of a variety of systemic and organ-specific autoimmune diseases map nonrandomly into eighteen chromosomal clusters, possibly explaining the occur-rence of several autoimmune diseases in a given individual or family. For example, Hashimoto’s thyroiditis is associated with a variety of organ-specific (e.g., TID, per-nicious anemia, autoimmune hepatitis, and Addison’s disease) and systemic (e.g., lupus, RA, and Sjögren’s syndrome) auto-immune diseases. Pedigrees with more than one systemic autoimmune disorder are not unusual. Of course, shared envi-ronmental influences could also explain familial clustering.
Among candidate genes, the MHC class II molecule is the most comprehen-sively studied. MHC polymorphisms are associated with development of RA, SLE, MS, TID, and other autoimmune diseases (Table 6.3). RA is a striking example. A shared epitope, consisting of a 5-amino acid sequence motif in the third allelic hypervariable region of the HLA-DRβ1 chain (QKRAA in the *0401 allele, QRRAA in the *0404 and *0101 alleles), is carried by 90 percent of patients with RA and is associated with disease severity. In addi-tion to MHC-linked genes, mutations or genetic polymorphisms involving non-MHC genes also are strong candidates for autoimmune disease susceptibility genes (Table 6.4). These include genetic polymor-phisms or deficiency of molecules involved in the response to or clearance of immune complexes (e.g., C1q, C4, FcγRIIa, FcγRIIb, FcγRIIIa, mannose-binding lectin), which are associated with SLE, as well as genes influencing T-cell activation (PTPN22, CTLA-4), cytokine responses (STAT-4, IRF-5, Tyk2), or programmed cell death (Fas, PDCD1; Table 6.4).
Table 6.4 Some Candidate Non-MHC Autoimmune Disease Susceptibility Genes
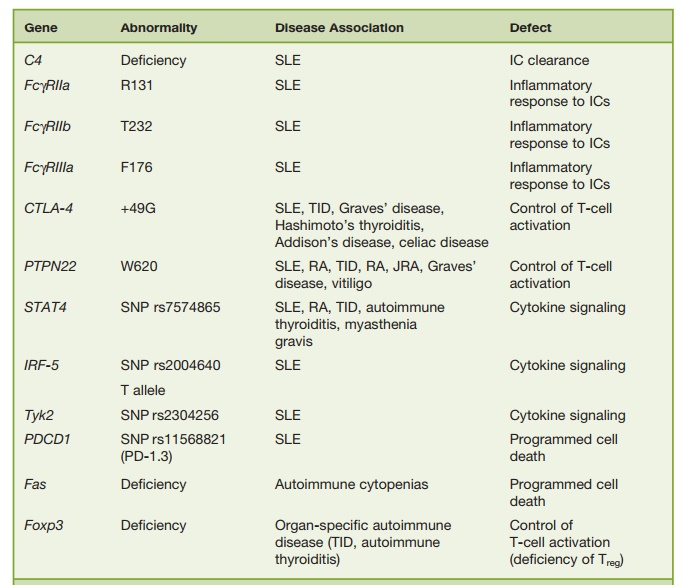
Many, if not most, systemic and organ-specific autoimmune diseases are thought to be multifactorial, involving multiple genetic defects consistent with the thresh-old liability model of multifactorial inheri-tance. This model supposes a continuously distributed genetically determined liability to the development of disease. Individuals who develop disease will bear multiple disease susceptibility genes. Because of the normal distribution of genes, first-degree relatives will have much higher risk of developing disease than the general population, second-degree relatives will have a moderate risk, and third-degree relatives will have low risk.
Environmental Triggers of Autoimmune Disease
Environmental factors can trigger autoim-mune disease in susceptible hosts, as illus-trated by the initiation of disease by sun exposure in a subset of lupus patients. The wavelengths most likely to induce lupus fall in the UV range: UVC (200–290 nm), UVB (290–320 nm), and UVA (320–400 nm). UVB irradiation is mostly absorbed in the upper layers of the epidermis, whereas the longer wavelength UVA is able to reach the dermis. UV exposure can induce apop-tosis and release of immune mediators and activation of resident dendritic cells and T cells. Expression of certain self-antigens, such as Ro60/Ro52, on the surface of the apoptotic cells may lead to antibody-medi-ated inflammatory responses that could play a role in the pathogenesis of skin rashes in lupus.
The importance of environmental fac-tors is further illustrated by the induc-tion of a murine lupus syndrome by the hydrocarbon pristane (see “Animal Mod-els of Autoimmune Disease”), which appears to act, in part, through the induc-tion of type I interferon (IFN-α and IFN-β) production. Many other chemicals and drugs have been implicated as triggers of autoimmunity or autoimmune disease. Procainamide, hydralazine, chlorproma-zine, methyldopa, quinidine, minocycline, and nitrofurantoin all have been associ-ated with the induction of ANAs and in some cases antineutrophil cytoplasmic antibodies as well as in the pathogenesis of “drug-induced lupus,” most frequently manifested by serositis (inflammation of the pleura or pericardium) and arthri-tis. Silica is recognized as a precipitating factor for scleroderma, cigarette smoke may aggravate RA, and trichloroethyl-ene is thought to promote lupus in ani-mal models and possibly humans. Other chemical agents implicated in the patho-genesis of autoimmunity include heavy metals such as mercury, gold, and cadmium, pesticides, herbici des, hydrazine, and certain dyes.
Infections also are implicated in the pathogenesis of autoimmune disease. The classic example is rheumatic fever, which is thought to be a consequence of cross-reactivity or “molecular mim-icry” between antigens carried by certain strains of streptococci and self-antigens of the heart. Mycoplasma pneumonia can induce the production of cold agglutinins, polyclonal cold-reactive IgM autoantibod-ies against the erythrocyte I, or i antigens that can cause complement-mediated AIHA (see “Type II Autoimmune Reactions”). A variety of other parasitic
(e.g., schistosomiasis, Chagas’ disease), bacterial (e.g., Helicobacter pylori, staphy-lococci, salmonella, Lyme borreliosis), mycobacterial (e.g., tuberculosis, leprosy), and viral (e.g., cytomegalovirus, Epstein-Barr virus, hepatitis C, Coxsackie virus, parvovirus B19) infections can be compli-cated by autoimmunity. Proposed mecha-nisms include molecular mimicry and the chronic overproduction of cytokines, such as IFN-α. Indeed, therapy with IFN-α can lead to the development of autoimmune diseases such as autoimmune thyroiditis and SLE
Maintenance of Self-Tolerance
Environmental triggers, such as sunlight, drugs/chemicals, and infectious agents, act on a genetic background that regulates tolerance to self. The immune system has evolved a remarkable ability to distin-guish self from nonself. Immune tolerance is achieved by multiple mechanisms, operating both centrally and peripher-ally. Central tolerance occurs during the development of T and B lymphocytes in the thymus and bone marrow, respec-tively. This mostly involves the deletion of autoreactive cells before they exit the primary lymphoid organs. In general, lymphocytes exhibiting strong reactiv-ity for ubiquitously expressed self-anti-gens are deleted in this manner, whereas autoreactive cells of lower affinity for self may escape central tolerance. These cells are held in check by peripheral tolerance mechanisms. Peripheral tolerance is medi-ated by deletion, anergy, and suppression as well as by “neglect” or “ignorance,” acting on autoreactive lymphocytes after they exit the primary organs. In general, lymphocyte activation requires two sig-nals, one delivered by the antigen receptor (T-cell antigen receptor or surface immu-noglobulin) and second, a co-stimulatory signal. For T-cell activation, this co-stimu-latory signal is delivered by the interac-tion of molecules expressed on the surface of professional antigen-presenting cells or B cells, such as CD80 and CD86, which interact with CD28 (or other receptors) on the T-cell surface. In the case of B cells, the co-stimulatory signal is delivered by CD40 ligand, a surface protein expressed by activated helper T cells that interacts with CD40 on the surface of B lympho-cytes. In the absence of a co-stimulatory signal, engagement of the T- or B-cell anti- gen receptor leads to a state of anergy (the inability of the lymphocyte to respond to its specific antigen).
Many self-antigens are expressed at a very low level that is insufficient to induce T-cell activation. In the case of T cells, which recognize short peptides asso-ciated with MHC molecules, the induc-tion of self-tolerance requires the genera-tion of a sufficient amount of self-peptide in antigen-presenting cells to stimulate T-cell deletion or anergy. Self-peptides that are generated inefficiently by the anti-gen-presenting cells can neither stimulate immunity nor induce tolerance; that is, the immune system remains “ignorant” of them. If these minor self-peptides are pro-duced in larger amounts and exposed to the immune system in the presence of an inducer of co-stimulatory molecules (e.g., adjuvants), they have the capacity to stim-ulate an immune response. This has been shown experimentally with peptides gen-erated in vitro using proteolytic enzymes or using synthetic self-peptides.
Regulatory T cells (Treg) also play an important role in maintaining peripheral tolerance. Several different subsets of Treg have been reported, but one of the most intensely studied is the CD4+CD25+Foxp3+ subset, which represents about 10 percent of total CD4+ cells. These cells regulate T-cell activation by a cell–cell contact-dependent mechanism and through the secretion of inhibitory cytokines such as IL-10 and transforming growth factor-beta (TGFβ). They suppress both naïve and memory T-cell responses and down-regu-late the expression of pro-inflammatory cytokines and co-stimulatory molecules on the antigen-presenting cells. These cells are induced in an antigen-specific man-ner, but the subsequent suppressive effects are not antigen specific. Genetic defects in Foxp3, a transcription factor that is the key controller of Treg function, lead to organ-specific autoimmune or autoinflamma-tory diseases. The scurfy mouse has an X-linked defect of the Foxp3 gene that is lethal in males, which exhibit hyperactiva-tion of CD4+ T cells and overproduction of inflammatory cytokines. Foxp3 muta-tions in humans are the cause of the IPEX (immune dysregulation, polyendocrinopa-thy, enteropathy, X-linked syndrome) syn-drome. These individuals develop organ-specific autoimmune diseases, such as TID, autoimmune thyroiditis, and inflammatory bowel disease. Interestingly, however, the development of systemic autoimmune dis-ease is not part of the syndrome in either mice or humans.
Finally, antigen-presenting cells play an important role in the induction of tol-erance. Dendritic cells can either initiate T-cell activation and proliferation or promote peripheral tolerance through the deletion of autoreactive T cells, depending on their maturation state. Tolerance is induced when antigens are presented by immature dendritic cells and these cells also play a role in the generation and maintenance of Treg.
Related Topics