Chapter: Basic & Clinical Pharmacology : Agents Used in Dyslipidemia
The Primary Hypercholesterolemias
THE PRIMARY HYPERCHOLESTEROLEMIAS
Familial Hypercholesterolemia
Familial
hypercholesterolemia is an autosomal dominant trait. Although levels of LDL
tend to increase throughout childhood, the diagnosis can often be made on the
basis of elevated umbilical cord blood cholesterol. In most heterozygotes,
cholesterol levels range from 260 to 500 mg/dL. Triglycerides are usually
normal, tendon xanthomas are often present, and arcus corneae and xan-thelasma
may appear in the third decade. Coronary disease tends to occur prematurely. In
homozygous familial hypercholester-olemia, which can lead to coronary disease
in childhood, levels of cholesterol often exceed 1000 mg/dL and early tuberous
and ten-dinous xanthomas occur. These patients may also develop elevated
plaque-like xanthomas of the aortic valve, digital webs, buttocks, and
extremities.
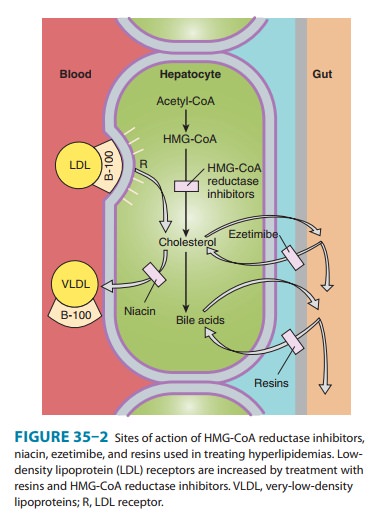
Defects
of LDL receptors underlie familial hypercholester-olemia. Some individuals have
combined heterozygosity for alleles producing nonfunctional and kinetically
impaired receptors. In heterozygous patients, LDL can be normalized with
combined drug regimens (Figure 35–2). Homozygotes and those with com-bined
heterozygosity whose receptors retain even minimal func-tion may partially
respond to niacin, ezetimibe, or reductase inhibitors.
Familial Ligand-Defective Apolipoprotein B-100
Defects in the domain of apo B-100 that binds to the LDL receptor impair the endocytosis of LDL, leading to hypercholes-terolemia of moderate severity. Tendon xanthomas may occur. These disorders are as prevalent as familial hypercholester-olemia. Response to reductase inhibitors is variable. Up-regulation of LDL receptors in liver increases endocytosis of LDL precursors but does not increase uptake of ligand-defective LDL particles. Niacin often has beneficial effects by reducing VLDL production.
Familial Combined Hyperlipoproteinemia
As
described, some persons with familial combined hyperlipopro-teinemia have only
an elevation in LDL. Serum cholesterol is usually less than 350 mg/dL. Dietary
and drug treatment, usually with a reductase inhibitor, is indicated. It may be
necessary to add niacin or ezetimibe to normalize LDL.
Lp(a) Hyperlipoproteinemia
This familial
disorder, which is associated with increased atherogen-esis, is determined
chiefly by alleles that dictate increased produc-tion of the (a) protein
moiety. Lp(a) can be secondarily elevated in patients with severe nephrosis and
certain other inflammatory states. Niacin reduces levels of Lp(a) in many
patients.
Other Disorders
Deficiency of
cholesterol 7α-hydroxylase
can increase LDL in the heterozygous state. Homozygotes can also have elevated
triglycerides, resistance to reductase inhibitors, and increased risk of
gallstones and coronary disease. Autosomal recessive hypercholesterolemia is
due to mutations in a protein that normally assists in endocytosis of LDL. Some
mutations in the PCSK9 gene also
cause isolated elevations of LDL. Niacin, ezetimibe, and reductase inhibitors
may be useful, vari-ably, in these disorders.
HDL Deficiency
Rare genetic disorders,
including Tangier disease and LCAT (lecithin:cholesterol acyltransferase)
deficiency, are associated with extremely low levels of HDL. Familial
hypoalphalipoproteinemia is a more common disorder with levels of HDL
cholesterol usually below 35 mg/dL in men and 45 mg/dL in women. These patients
tend to have premature atherosclerosis, and the low HDL may be the only
identified risk factor. Management should include special attention to
avoidance or treatment of other risk factors. Niacin increases HDL in many of
these patients. Reductase inhibitors and fibric acid derivatives exert lesser
effects.
In the presence of
hypertriglyceridemia, HDL cholesterol is low because of exchange of cholesteryl
esters from HDL into triglyceride-rich lipoproteins. Treatment of the
hypertriglyceri-demia may increase or normalize the HDL level.
Related Topics