Chapter: Basic & Clinical Pharmacology : Agents Used in Dyslipidemia
Competitive Inhibitors of HMG-COA Reductase (Reductase Inhibitors; 'Statins')
COMPETITIVE INHIBITORS OF HMG-COA
REDUCTASE (REDUCTASE INHIBITORS; “STATINS”)
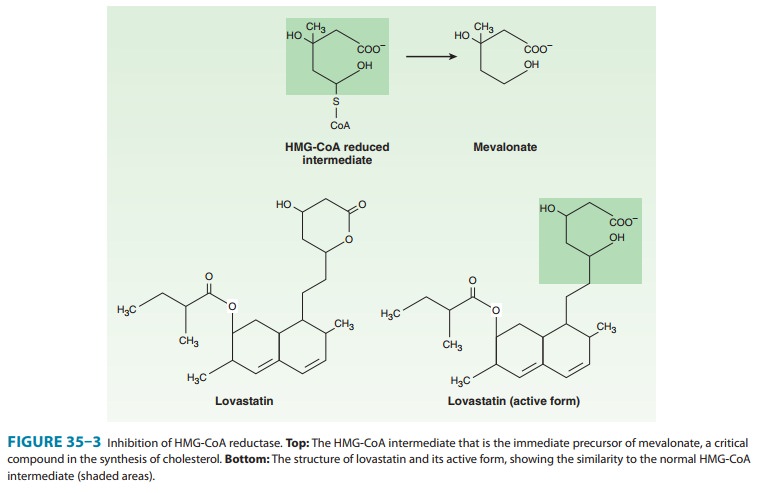
These compounds are structural analogs of HMG-CoA (3-hydroxy-3-methylglutaryl-coenzyme A, Figure 35–3). Lovastatin, atorvas-tatin, fluvastatin, pravastatin, simvastatin, rosuvastatin, and pitavastatin belong to this class. They are most effective in reduc-ing LDL. Other effects include decreased oxidative stress and vascular inflammation with increased stability of atherosclerotic lesions. It has become standard practice to initiate reductase inhibitor therapy immediately after acute coronary syndromes, regardless of lipid levels.
Chemistry & Pharmacokinetics
Lovastatin and
simvastatin are inactive lactone prodrugs that are hydrolyzed in the
gastrointestinal tract to the active β-hydroxyl derivatives, whereas pravastatin has
an open, active lactone ring. Atorvastatin, fluvastatin, and rosuvastatin are
fluorine-containing congeners that are active as given. Absorption of the
ingested doses of the reductase inhibitors varies from 40% to 75% with the
excep-tion of fluvastatin, which is almost completely absorbed. All have high
first-pass extraction by the liver. Most of the absorbed dose is excreted in
the bile; 5–20% is excreted in the urine. Plasma half-lives of these drugs
range from 1 to 3 hours except for atorvastatin (14 hours), pitavastatin (12
hours), and rosuvastatin (19 hours).
Mechanism of Action
HMG-CoA reductase
mediates the first committed step in sterol biosynthesis. The active forms of
the reductase inhibitors are struc-tural analogs of the HMG-CoA intermediate
(Figure 35–3) that is formed by HMG-CoA reductase in the synthesis of
mevalonate. These analogs cause partial inhibition of the enzyme and thus may
impair the synthesis of isoprenoids such as ubiquinone and dolichol and the
prenylation of proteins. It is not known whetherthis has biologic significance.
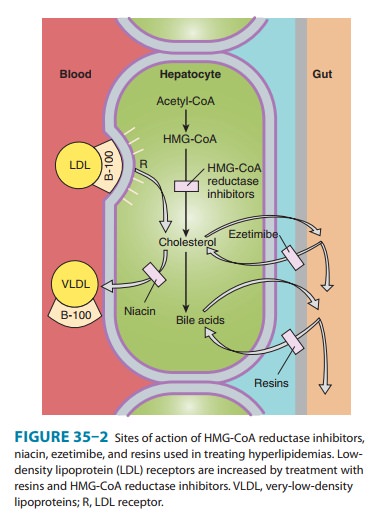
However, the reductase inhibitors clearly induce an increase in high-affinity
LDL receptors. This effect increases both the fractional catabolic rate of LDL
and the liver’s extraction of LDL precursors (VLDL remnants) from the blood,
thus reducing LDL (Figure 35–2). Because of marked first-pass hepatic
extraction, the major effect is on the liver. Preferential activity in liver of
some congeners appears to be attributable to tissue-specific differences in
uptake. Modest decreases in plasma triglycerides and small increases in HDL
also occur.
Clinical trials
involving many of the statins have demonstrated significant reduction of new
coronary events and atherothrom-botic stroke. Mechanisms other than reduction
of lipoprotein levels appear to be involved. The availability of isoprenyl
groups from the HMG-CoA pathway for prenylation of proteins is reduced by
statins, resulting in reduced prenylation of Rho and Rab proteins. Prenylated
Rho activates Rho kinase, which medi-ates a number of mechanisms in vascular
biology. The observation that reduction in new coronary events occurs more
rapidly than changes in morphology of arterial plaques suggests that these
pleiotropic effects may be important. Likewise, decreased prenyla-tion of Rab
reduces the accumulation of Aβ protein in neurons, possibly mitigating the
manifestations of Alzheimer’s disease.
Therapeutic Uses & Dosage
Reductase inhibitors
are useful alone or with resins, niacin, or ezetimibe in reducing levels of
LDL. Women with hyperlipidemia who are pregnant, lactating, or likely to become
pregnant should not be given these agents. Use in children is restricted to
selected patients with familial hypercholesterolemia or familial combined
hyperlipidemia.
Because cholesterol
synthesis occurs predominantly at night, reductase inhibitors—except
atorvastatin and rosuvastatin— should be given in the evening if a single daily
dose is used. Absorption generally (with the exception of pravastatin) is
enhanced by food. Daily doses of lovastatin vary from 10 to 80 mg. Pravastatin
is nearly as potent on a mass basis as lovastatin with a maximum recommended
daily dose of 80 mg. Simvastatin is twice as potent and is given in doses of 5–80
mg daily. Because of increased risk of myopathy with the 80 mg/day dose, the
FDA issued labelling for scaled dosing of simvastatin and Vytorin in June 2011.
Fluvastatin appears to be about half as potent as lovas-tatin on a mass basis
and is given in doses of 10–80 mg daily. Atorvastatin is given in doses of
10–80 mg/d, and rosuvastatin, the most efficacious agent for severe
hypercholesterolemia, at 5–40 mg/d. The dose-response curves of pravastatin and
especially of fluvastatin tend to level off in the upper part of the dosage
range in patients with moderate to severe hypercholesterolemia. Those of other
statins are somewhat more linear.
Toxicity
Elevations of serum
aminotransferase activity (up to three times normal) occur in some patients.
This is often intermittent and usually not associated with other evidence of
hepatic toxicity. Therapy may be continued in such patients in the absence of
symptoms if aminotransferase levels are monitored and stable. In some patients,
who may have underlying liver disease or a history of alcohol abuse, levels may
exceed three times normal. This find-ing portends more severe hepatic toxicity.
These patients may present with malaise, anorexia, and precipitous decreases in
LDL. Medication should be discontinued immediately in these patients and in
asymptomatic patients whose aminotransferase activity is persistently elevated
to more than three times the upper limit of normal. These agents should be used
with caution and in reduced dosage in patients with hepatic parenchymal
disease, Asians, and the elderly. Severe hepatic disease may preclude their
use. In gen-eral, aminotransferase activity should be measured at baseline, at
1–2 months, and then every 6–12 months (if stable). Monitoring of liver enzymes
should be more frequent if the patient is taking other drugs that have
potential interactions with the statin. Fasting plasma glucose levels tend to
increase 5–7 mg/dL with statin treatment.
Minor increases in
creatine kinase (CK) activity in plasma are observed in some patients receiving
reductase inhibitors, frequently associated with heavy physical activity.
Rarely, patients may have marked elevations in CK activity, often accompanied
by general-ized discomfort or weakness in skeletal muscles. If the drug is not
discontinued, myoglobinuria can occur, leading to renal injury. Myopathy may
occur with monotherapy, but there is an increased incidence in patients also
receiving certain other drugs. Genetic variation in an anion transporter
(OATP1B1) is associated with severe myopathy and rhabdomyolysis induced by
statins.
The catabolism of
lovastatin, simvastatin, and atorvastatin proceeds chiefly through CYP3A4,
whereas that of fluvastatin and rosuvastatin, and to a lesser extent
pitavastatin, is mediated by CYP2C9. Pravastatin is catabolized through other
pathways, including sulfation. The 3A4-dependent reductase inhibitors tend to
accumulate in plasma in the presence of drugs that inhibit or compete for the
3A4 cytochrome. These include the macrolide antibiotics, cyclosporine, ketoconazole
and its congeners, some HIV protease inhibitors, tacrolimus, nefazodone,
fibrates, parox-etine, venlafaxine, and others . Concomitant use of reductase
inhibitors with amiodarone or verapamil also causes an increased risk of
myopathy.
Conversely, drugs such
as phenytoin, griseofulvin, barbiturates, rifampin, and thiazolidinediones
increase expression of CYP3A4 and can reduce the plasma concentrations of the
3A4-dependent reductase inhibitors. Inhibitors of CYP2C9 such as ketoconazole
and its congeners, metronidazole, sulfinpyrazone, amiodarone, and cimetidine
may increase plasma levels of fluvastatin and rosuvasta-tin. Pravastatin and
rosuvastatin appear to be the statins of choice for use with verapamil, the
ketoconazole group of antifungal agents, macrolides, and cyclosporine. Doses
should be kept low and the patient monitored frequently. Plasma levels of
lovastatin, simvastatin, and atorvastatin may be elevated in patients ingesting
more than 1 liter of grapefruit juice daily. All statins undergo
gly-cosylation, thus creating an interaction with gemfibrozil.
Creatine kinase
activity should be measured in patients receiving potentially interacting drug
combinations. In all patients, CK should be measured at baseline. If muscle
pain, tenderness, or weak-ness appears, CK should be measured immediately and
the drug discontinued if activity is elevated significantly over baseline. The
myopathy usually reverses promptly upon cessation of therapy. If the
association is unclear, the patient can be rechallenged under close
surveillance. Myopathy in the absence of elevated CK can occur. Rarely,
hypersensitivity syndromes have been reported that include a lupus-like
disorder and peripheral neuropathy.Reductase inhibitors should be temporarily
discontinued in the event of serious illness, trauma, or major surgery.
Related Topics