Chapter: Basic & Clinical Pharmacology : The Eicosanoids:Prostaglandins, Thromboxanes, Leukotrienes, & Related Compounds
Synthesis of Eicosanoids
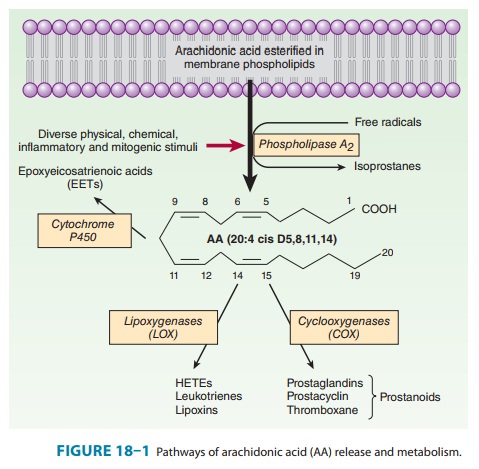
SYNTHESIS OF EICOSANOIDS
Products of Prostaglandin Endoperoxide Synthases (Cyclooxygenases)
Two
unique COX isozymes convert AA into prostaglandin endoperoxides. PGH synthase-1
(COX-1) is expressed constitu-tively
in most cells. In contrast, PGH synthase-2 (COX-2)
is inducible; its expression varies depending on the stimulus. COX-2 is an
immediate early-response gene product that is markedlyup-regulated by shear
stress, growth factors, tumor promoters, and cytokines. COX-1 generates
prostanoids for “housekeeping” such as gastric epithelial cytoprotection,
whereas COX-2 is the major source of prostanoids in inflammation and cancer.
This distinc-tion is overly simplistic, however; there are both physiologic and
pathophysiologic processes in which each enzyme is uniquely involved and others
in which they function coordinately. For example, endothelial COX-2 is the
primary source of vascular prostacyclin (PGI2), whereas renal COX-2-derived
prostanoids are important for normal renal development and maintenance of
function. Nonsteroidal anti-inflammatory drugs (NSAIDs;) exert their
therapeutic effects through inhibition of the COXs. Indomethacin and sulindac
are slightly selective for COX-1. Meclofenamate and ibuprofen are approximately
equipo-tent on COX-1 and COX-2, whereas celecoxib = diclofenac < rofecoxib = lumiracoxib < etoricoxib in inhibition of COX-2 (listed in
order of increasing average selectivity). Aspirin acetylates and inhibits both
enzymes covalently. Low doses (< 100 mg/d) inhibit preferentially, but not
exclusively, platelet COX-1, whereas higher doses inhibit both systemic COX-1
and COX-2.
Both COX-1 and COX-2 promote the uptake of two mole-cules of oxygen by cyclization of arachidonic acid to yield a C9–C11 endoperoxide C15 hydroperoxide (Figure 18–2). This product is PGG2, which is then rapidly modified by the peroxidase moiety of the COX enzyme to add a 15-hydroxyl group that is essential for biologic activity. This product is PGH2. Both endoperoxides are highly unstable. Analogous families—PGH1 and PGH3 and all their subsequent products—are derived from homo-γ-linolenic acid and eicosapentaenoic acid, respectively.
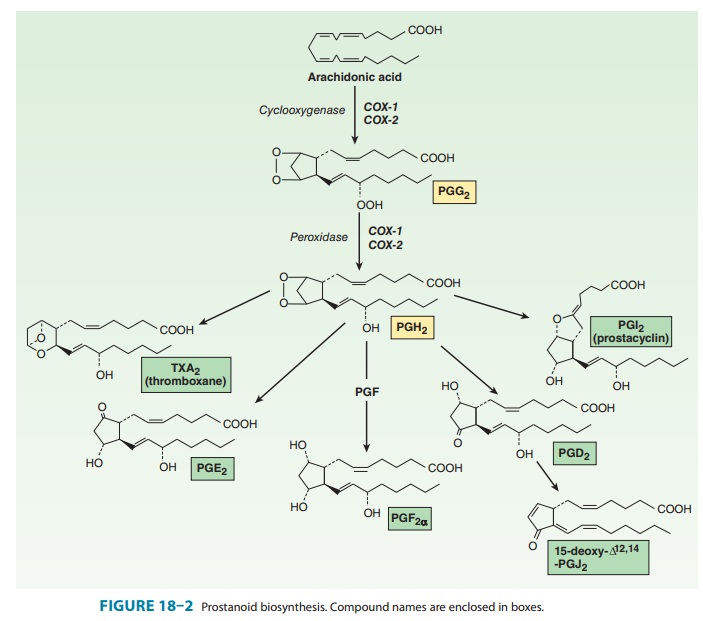
The
prostaglandins, thromboxane, and prostacyclin, collec-tively termed the
prostanoids, are generated from PGH2 through the action of
downstream isomerases and synthases. These termi-nal enzymes are expressed in a
relatively cell-specific fashion, such that most cells make one or two dominant
prostanoids. The pros-taglandins differ from each other in two ways: (1) in the
substitu-ents of the pentane ring (indicated by the last letter, eg, E and F in
PGE and PGF) and (2) in the number of double bonds in the side chains
(indicated by the subscript, eg, PGE1, PGE2). PGH2
is metabolized by prostacyclin, thromboxane, and PGF synthases (PGIS, TXAS, and
PGFS) to PGI2, TXA2, and PGF2α, respec-tively. Two
additional enzymes, 9,11-endoperoxide reductase and 9-ketoreductase, provide
for PGF2α synthesis from PGH2 and PGE2,
respectively. At least three PGE 2 synthases have been iden-tified:
microsomal (m) PGES-1, the more readily inducible mPGES-2, and cytosolic PGES.
There are two distinct PGDS isoforms, the lipocalin-type PGDS and the
hematopoietic PGDS.Several products of the arachidonate series are of current
clini-cal importance. Alprostadil
(PGE1) may be used for its smooth muscle relaxing effects to
maintain the ductus arteriosus patent in some neonates awaiting cardiac surgery
and in the treatment of impotence. Misoprostol,
a PGE1 derivative, is a cytoprotective prostaglandin used in
preventing peptic ulcer and in combination with mifepristone (RU-486) for
terminating early pregnancies. PGE2and PGF2are used in obstetrics
to induce labor. Latanoprost and
several similar compounds are topically activePGF2α derivatives used in
ophthalmology to treat open-angle glaucoma. Prostacyclin (PGI2, epoprostenol) is synthesized mainly
by the vascular endothelium and is a powerful vasodilator and inhibitor of
platelet aggregation. It is used clinically to treat pulmonary hypertension and
portopulmonary hypertension. In contrast, thromboxane
(TXA2) has undesirable properties (aggre-gation of platelets,
vasoconstriction). Therefore TXA2-receptor antagonists and synthesis
inhibitors have been developed for car-diovascular indications, although these
(except for aspirin) have yet to establish a place in clinical usage. All the
naturally occurring COX products undergo rapid metabolism to inactive products
either by hydration (for PGI 2 and TXA2) or by oxidation
of the key 15-hydroxyl group to the cor-responding ketone by prostaglandin
15-hydroxy prostaglandin dehydrogenase (15-PGDH) after cellular uptake via an
organic anion transporter polypeptide (OATP 2A1). Further metabolism is by 13
reduction, β-oxidation,
and ω-oxidation.
The inactive metabolites can be determined in blood and urine by immunoassay or
mass spectrometry as a measure of the in vivo synthesis of their parent
compounds.
Products of Lipoxygenase
The
metabolism of AA by the 5-, 12-, and
15-lipoxygenases (LOX) results in the production of
hydroperoxyeicosatetraenoic acids (HPETEs), which rapidly convert to hydroxy
derivatives (HETEs) and leukotrienes (Figure 18–3). The most actively
investigated leu-kotrienes are those produced by the 5-LOX present in
leukocytes (neutrophils, basophils, eosinophils, and monocyte-macrophages) and
other inflammatory cells such as mast cells and dendritic cells. This pathway
is of great interest since it is associated with asthma, anaphylactic shock,
and cardiovascular disease. Stimulation of these cells elevates intracellular
Ca2+ and releases arachidonate; incorporation of molecular oxygen by
5-LOX, in association with 5-LOX-activating
protein (FLAP), then yields the unstableepoxide leukotriene A4
(LTA4). This intermediate is either con-verted to the dihydroxy
leukotriene B4 (LTB4), via the action of LTA4
hydrolase, or is conjugated with glutathione to yield leukot-riene C4
(LTC4), by LTC4 synthase. Sequential degradation of the
glutathione moiety by peptidases yields LTD4 and LTE4.
These three products, LTC4, D4, and E4, are
called cysteinyl leukot-rienes. Although leukotrienes are predominantly
generated in leukocytes, non-leukocyte cells (eg, endothelial cells) that
express enzymes downstream of 5-LOX/FLAP can take up and convert
leukocyte-derived LTA4 in a process termed transcellular
biosyn-thesis. Transcellular formation of prostaglandins has also been shown;
for example, endothelial cells can use platelet PGH2 to form PGI2.
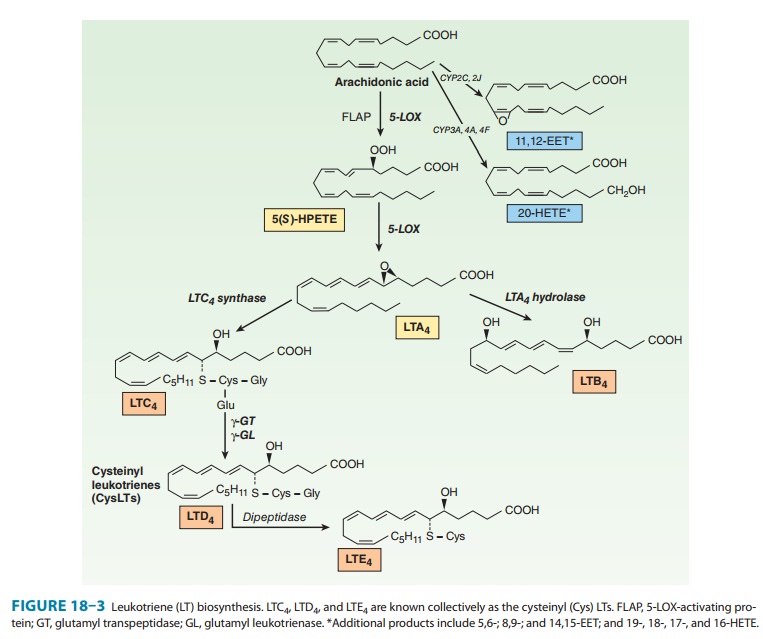
LTC4and LTD4are
potent bronchoconstrictors and are recog-nized as the primary components of the
slow-reacting substanceof anaphylaxis
(SRS-A) that is secreted in asthma and anaphy-laxis. There are four current
approaches to antileukotriene drug development: 5-LOX enzyme inhibitors,
leukotriene-receptor antagonists, inhibitors of FLAP, and phospholipase A2
inhibitors.
LTA4,
the primary product of 5-LOX, can be converted with appropriate stimulation via
12-LOX in platelets to the lipoxins
LXA4 and LXB4 in vitro. These mediators can also be
generated through 5-LOX metabolism of 15-HETE, the product of 15-LOX-2
metabolism of arachidonic acid. 15-LOX-1 prefers linoleic acid as a substrate
forming 15S-hydroxyoctadecadienoic acid. The stereochemical isomer, 15( R )-HETE, may be derived from the action
of aspirin-acetylated COX-2 and further trans-formed in leukocytes by 5-LOX to
15-epi-LXA4 or 15-epi-LXB4, the so-called
aspirin-triggered lipoxins. Synthetic lipoxins and epi-lipoxins exert anti-inflammatory
actions when applied in vivo. Although these compounds can be formed from
endoge-nous substrates in vitro and when synthesized may have potent biologic
effects, the importance of the endogenous compounds in vivo in human biology
remains ill defined. 12-HETE, a product of 12-LOX, can also undergo a catalyzed
molecular rearrange-ment to epoxyhydroxyeicosatrienoic acids called hepoxilins. Proinflammatory effects of
synthetic hepoxilins have been reported although their biologic relevance is
unclear.
The
LOXs located in epidermal cells are distinct from “conven-tional”
enzymes—arachidonic acid and linoleic acid are appar-ently not the natural
substrates for epidermal LOX. Epidermal accumulation of 12(R )-HETE is a feature of psoriasis and ich-thyosis and inhibitors
of 12(R )-LOX are under investigation
for the treatment of these proliferative skin disorders.
Epoxygenase Products
Specific
isozymes of microsomal cytochrome P450 monooxyge-nases convert AA to hydroxy-
or epoxyeicosatrienoic acids (Figures 18–1 and 18–3). The products are 20-HETE,
generated by the CYP hydroxylases (CYP3A, 4A, 4F) and the 5,6-, 8,9-, 11,12-,
and 14,15-epoxyeicosatrienoic acids (EETs), which arise from the CYP
epoxygenase (2J, 2C). Their biosynthesis can be altered by pharmacologic,
nutritional, and genetic factors that affect P450 expression. The biologic
actions of the EETs are reduced by their conversion to the corresponding, and
biologically less active, dihy-droxyeicosatrienoic acids (DHETs) through the
action of soluble epoxide hydrolase (sEH). Unlike the prostaglandins, the EETs
can be esterified into phospholipids, which then act as storage sites.
Intracellular fatty acid-binding proteins promote EET uptake into cells,
incorporation into phospholipids, and availability to sEH. EETs are synthesized
in endothelial cells and cause vasodilation in a number of vascular beds by
activating the smooth muscle large conductance Ca2+-activated K+
channels. This results in smooth muscle cell hyperpolarization and
vasodilation, leading to reduced blood pressure. Substantial evidence indicates
that EETs may function as endothelium-derived
hyperpolarizing factors, par-ticularly in the coronary circulation.
Consequently there is interest in inhibitors of soluble sEH as potential antithrombotic
and antihypertensive drugs. An exception to the general response to EETs as
vasodilators is the pulmonary vasculature where they cause vasoconstriction. It
is unclear yet whether this activity of EETs may limit the potential clinical
use of sEH inhibitors. Down-regulation of pulmonary sEH may contribute to
pulmonary hypertension. Anti-inflammatory, antiapoptotic, and proangio-genic
actions of the EETs have also been reported.
Isoeicosanoids
The
isoeicosanoids, a family of eicosanoid isomers, are formed nonenzymatically by
direct free radical-based action on AA and related lipid substrates.
Isoprostanes are prostaglandin stereoiso-mers. Because prostaglandins have many
asymmetric centers, they have a large number of potential stereoisomers. COX is
not needed for the formation of the isoprostanes, and its inhibition with
aspirin or other NSAIDs should not affect the isoprostane pathway. The primary
epimerization mechanism is peroxidation of arachidonate by free radicals.
Peroxidation occurs while arachi-donic acid is still esterified to the membrane
phospholipids. Thus, unlike prostaglandins, these stereoisomers are “stored” as
part of the membrane. They are then cleaved by phospholipases, circu-late, and
are excreted in urine. Isoprostanes are present in relatively large amounts
(tenfold greater in blood and urine than the COX-derived prostaglandins). They
have potent vasoconstrictor effects when infused into renal and other vascular
beds and may activate prostanoid receptors. They also may modulate other
aspects of vascular function, including leukocyte and platelet adhesive
inter-actions and angiogenesis. It has been speculated that they may contribute
to the pathophysiology of inflammatory responses in a manner insensitive to COX
inhibitors. A particular difficulty in assessing the likely biologic functions
of isoprostanes—several of which have been shown to serve as incidental ligands
at prosta-glandin receptors—is that while high concentrations of individual
isoprostanes may be necessary to elicit a response, multiple com-pounds are
formed coincidentally in vivo under conditions of oxidant stress. Analogous
leukotriene and EET isomers have been described.
Related Topics