Chapter: Basic & Clinical Pharmacology : The Eicosanoids:Prostaglandins, Thromboxanes, Leukotrienes, & Related Compounds
Clinical Pharmacology of Eicosanoids
CLINICAL
PHARMACOLOGY OF EICOSANOIDS
Several
approaches have been used in the clinical application of eicosanoids. First,
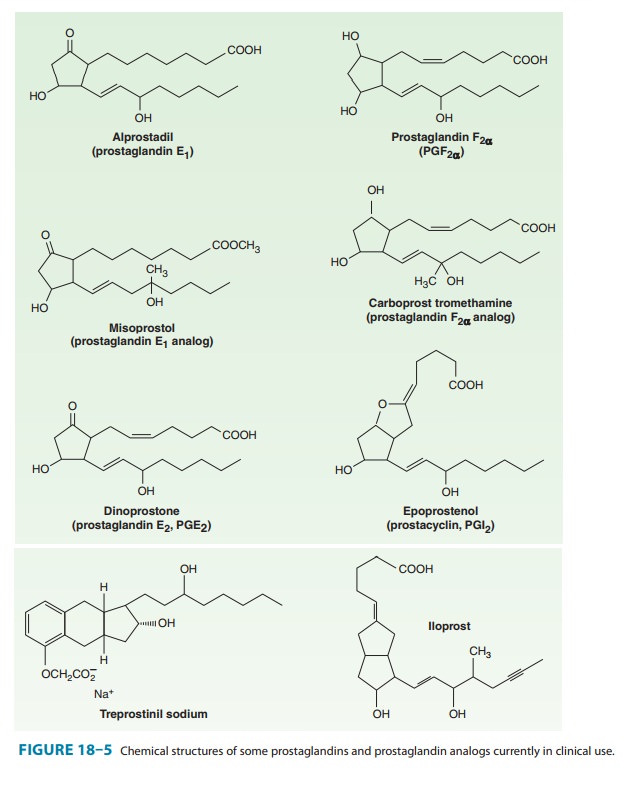
stable oral or parenteral long-acting analogs of the naturally occurring
prostaglandins have been developed (Figure 18–5). Second, enzyme inhibitors and
receptor antago-nists have been developed to interfere with the synthesis or
effects of the eicosanoids. The discovery of COX-2 as a major source of
inflammatory prostanoids led to the development of selective COX-2 inhibitors
in an effort to preserve the gastrointestinal and renal functions directed
through COX-1, thereby reducing toxic-ity. However, it is apparent that the
marked decrease in biosynthe-sis of PGI2 that follows COX-2
inhibition occurring without a concurrent inhibition of platelet COX-1-derived
TXA2 removes a protective constraint on endogenous mediators of cardiovascular
dysfunction and leads to an increase in cardiovascular events in patients
taking selective COX-2 inhibitors. Third, efforts at dietary manipulation—to
change the polyunsaturated fatty acid precursors in the cell membrane
phospholipids and so change eicosanoid synthesis—is used extensively in
over-the-counter products and in diets emphasizing increased consumption of
cold-water fish.
Female Reproductive System
Studies
with knockout mice have confirmed a role for prostaglan-dins in reproduction
and parturition. COX-1-derived PGF2α appears important for luteolysis, consistent
with delayed parturi-tion in COX-1-deficient mice. A complex interplay between
PGF2α and oxytocin is critical to the onset of labor. EP2
receptor-deficient mice demonstrate a preimplantation defect, which under-lies
some of the breeding difficulties seen in COX-2 knockouts.
A. Abortion
PGE2
and PGF2α have potent oxytocic actions. The ability of
the E and F prostaglandins and their analogs to terminate pregnancy at any
stage by promoting uterine contractions has been adapted to common clinical
use. Many studies worldwide have estab-lished that prostaglandin administration
efficiently terminatespregnancy. The drugs are used for first- and
second-trimester abortion and for priming or ripening the cervix before abortion.
These prostaglandins appear to soften the cervix by increasing proteoglycan
content and changing the biophysical properties of collagen.
Dinoprostone, a synthetic preparation of PGE2, is
adminis-tered vaginally for oxytocic use. In the USA, it is approved for
inducing abortion in the second trimester of pregnancy, for missed abortion,
for benign hydatidiform mole, and for ripen-ing of the cervix for induction of
labor in patients at or near term.
Dinoprostone
stimulates the contraction of the uterus through-out pregnancy. As the
pregnancy progresses, the uterus increases its contractile response, and the
contractile effect of oxytocin is potentiated as well. Dinoprostone also
directly affects the collage-nase of the cervix, resulting in softening. The vaginal
dose enters the maternal circulation, and a small amount is absorbed directly
by the uterus via the cervix and the lymphatic system. Dinoprostone is
metabolized in local tissues and on the first pass through the lungs (about
95%). The metabolites are mainly excreted in the urine. The plasma half-life is
2.5–5 minutes.For the induction of labor, dinoprostone is used either as a gel
(0.5
mg PGE2) or as a controlled-release formulation (10 mg PGE2)
that releases PGE2 in vivo at a rate of about 0.3 mg/h over 12
hours. An advantage of the controlled-release formulation is a lower incidence
of gastrointestinal side effects (< 1%).For abortifacient purposes, the recommended dosage is a
20-mg dinoprostone vaginal suppository repeated at 3- to 5-hour intervals
depending on the response of the uterus. The mean time to abortion is 17 hours,
but in more than 25% of cases, the abor-tion is incomplete and requires
additional intervention.
For
softening of the cervix at term, the preparations used are either a single
vaginal insert containing 10 mg PGE2 or a vaginal gel containing 0.5
mg PGE2 administered every 6 hours. The softening of the cervix for
induction of labor substantially short-ens the time to onset of labor and the
delivery time.
Antiprogestins
(eg, mifepristone) have been
combined with an oral oxytocic synthetic analog of PGE1 (misoprostol) to produce early abortion.
This regimen is available in the USA and Europe . The ease of use and the
effectiveness of the com-bination have aroused considerable opposition in some
quarters. The major toxicities are cramping pain and diarrhea. The oral and
vaginal routes of administration are equally effective, but the vaginal route
has been associated with an increased incidence of sepsis, so the oral route is
now recommended.
An analog of PGF2α is also used in obstetrics. This drug, car-boprost tromethamine (15-methyl-PGF2α; the 15-methyl groupprolongs the duration of action) is used to induce second-trimester abortions and to control postpartum hemorrhage that is not responding to conventional methods of management. The success rate is approximately 80%. It is administered as a single 250-mcg intramuscular injection, repeated if necessary. Vomiting and diar-rhea occur commonly, probably because of gastrointestinal smooth muscle stimulation. In some patients transient bronchoconstric-tion can occur. Transient elevations in temperature are seen in approximately one eighth of patients.
B. Facilitation of Labor
Numerous
studies have shown that PGE2, PGF2α, and their ana-logs
effectively initiate and stimulate labor, but PGF2α is one tenth as
potent as PGE2. There appears to be no difference in the efficacy of
PGE2 and PGF2α when they are administered intravenously;
however, the most common usage is local application of PGE2 analogs
(dinoprostone) to promote labor through ripening of the cervix. These agents
and oxytocin have similar success rates and comparable induction-to-delivery
intervals. The adverse effects of the prostaglandins are moderate, with a
slightly higher incidence of nausea, vomiting, and diarrhea than that produced
by oxytocin.
PGF2α has more
gastrointestinal toxicity than PGE2. Neither drug has significant
maternal cardiovascular toxicity in the recom-mended doses. In fact, PGE2
must be infused at a rate about 20 times faster than that used for induction of
labor to decrease blood pressure and increase heart rate. PGF2α is a
bronchoconstrictor and should be used with caution in women with asthma;
however, neither asthma attacks nor bronchoconstriction have been observed
during the induction of labor. Although both PGE2 and PGF2α pass the
fetoplacental barrier, fetal toxicity is uncommon.
The
effects of oral PGE2 administration (0.5–1.5 mg/h) have been
compared with those of intravenous oxytocin and oral demoxytocin, an oxytocin derivative, in the
induction of labor. Oral PGE2 is superior to the oral oxytocin
derivative and in most studies is as efficient as intravenous oxytocin. Oral
PGF2α causes too much gastrointestinal toxicity to be useful by this
route.
Theoretically,
PGE2 and PGF2α should be superior to oxytocin for inducing
labor in women with preeclampsia-eclampsia or car-diac and renal diseases
because, unlike oxytocin, they have no antidiuretic effect. In addition, PGE2
has natriuretic effects. However, the clinical benefits of these effects have
not been docu-mented. In cases of intrauterine fetal death, the prostaglandins
alone or with oxytocin seem to cause delivery effectively.
C. Dysmenorrhea
Primary
dysmenorrhea is attributable to increased endometrial syn-thesis of PGE2
and PGF2α during menstruation, with contractions of the
uterus that lead to ischemic pain. NSAIDs successfully inhibit the formation of
these prostaglandins and so relieve
dysmenorrhea in 75–85% of cases. Some of these drugs are available over the
counter. Aspirin is also effective in dysmenorrhea, but because it has low
potency and is quickly hydrolyzed, large doses and frequent administration are
necessary. In addition, the acetyla-tion of platelet COX, causing irreversible
inhibition of platelet TXA2 synthesis, may increase the amount of
menstrual bleeding.
Male Reproductive System
Intracavernosal
injection or urethral suppository therapy with alpros-tadil (PGE1) is a second-line treatment for
erectile dysfunction. Dosesof 2.5–25 mcg are used. Penile pain is a frequent
side effect, which may be related to the algesic effects of PGE derivatives;
however, only a few patients discontinue the use because of pain. Prolonged
erection and priapism are side effects that occur in less than 4% of patients
and are minimized by careful titration to the minimal effective dose. When
given by injection, alprostadil may be used as monotherapy or in combination
with either papaverine or phentolamine.
Renal System
Increased
biosynthesis of prostaglandins has been associated with one form of Bartter’s
syndrome. This is a rare disease characterized by low-to-normal blood pressure,
decreased sensitivity to angio-tensin, hyperreninemia, hyperaldosteronism, and
excessive loss of K+. There also is an increased excretion of
prostaglandins, espe-cially PGE metabolites, in the urine. After long-term
administra-tion of COX inhibitors, sensitivity to angiotensin, plasma renin
values, and the concentration of aldosterone in plasma return to normal.
Although plasma K+ rises, it remains low, and urinary wasting of K+
persists. Whether an increase in prostaglandin bio-synthesis is the cause of
Bartter’s syndrome or a reflection of a more basic physiologic defect is not
yet known.
Cardiovascular System
The
vasodilator effects of PGE compounds have been studied extensively in
hypertensive patients. These compounds also pro-mote sodium diuresis. Practical
application will require derivatives with oral activity, longer half-lives, and
fewer adverse effects.
A. Pulmonary Hypertension
PGI2
lowers peripheral, pulmonary, and coronary vascular resistance. It has been
used to treat primary pulmonary hypertension as well as sec-ondary pulmonary
hypertension, which sometimes occurs after mitral valve surgery. In addition,
prostacyclin has been used successfully to treat portopulmonary hypertension,
which arises secondary to liver disease. The first commercial preparation of
PGI2 (epoprostenol)
approved for treatment of primary pulmonary hypertension improves symptoms,
prolongs survival, and delays or prevents the need for lung or lung-heart
transplantation. Side effects include flushing, headache, hypotension, nausea,
and diarrhea. The extremely short plasma half-life (3–5 minutes) of
epoprostenol necessitates continuous intravenous infusion through a central line
for long-term treatment, which is its greatest limitation. Several prostacyclin
analogs with longer half-lives have been developed and used clinically. Iloprost (half-life about 30 minutes)
is usually inhaled six to nine times per day, although it has been delivered by
intravenous administration outside the USA. Treprostinil (half-life about 4 hours) may be delivered by
subcutaneousor intravenous infusion.
B. Peripheral Vascular Disease
A
number of studies have investigated the use of PGE1 and PGI2
compounds in Raynaud’s phenomenon and peripheral arterial disease. However,
these studies are mostly small and uncontrolled, and these therapies do not
have an established place in the treat-ment of peripheral vascular disease.
C. Patent Ductus Arteriosus
Patency
of the fetal ductus arteriosus depends on COX-2-derived PGE2 acting
on the EP4 receptor. At birth, reduced PGE2 levels, a
consequence of increased PGE2 metabolism, allow ductus arteriosus
closure. In certain types of congenital heart disease (eg, transposition of the
great arteries, pulmonary atresia, pulmonary artery stenosis), it is important
to maintain the patency of the neonate’s ductus arteriosus until corrective
surgery can be carried out. This can be achieved with alprostadil (PGE1).
Like PGE2, PGE1 is a vasodilator and an inhibitor of
platelet aggregation, and it contracts uterine and intestinal smooth muscle.
Adverse effects include apnea, bradycardia, hypotension, and hyperpyrexia.
Because of rapid pulmonary clearance (the half-life is about 5–10 minutes in
healthy adults and neonates), the drug must be continuously infused at an
initial rate of 0.05–0.1 mcg/kg/min, which may be increased to 0.4 mcg/kg/min.
Prolonged treatment has been associated with ductal fragility and rupture.
In
delayed closure of the ductus arteriosus, COX inhibitors are often used to
inhibit synthesis of PGE2 and so close the ductus. Premature infants
in whom respiratory distress develops due to failure of ductus closure can be
treated with a high degree of suc-cess with indomethacin. This treatment often
precludes the need for surgical closure of the ductus.
Blood
As
noted above, eicosanoids are involved in thrombosis because TXA2
promotes platelet aggregation while PGI 2, and perhaps also PGE2
and PGD 2, are platelet antagonists. Chronic administration of
low-dose aspirin (81 mg/d) selectively and irreversibly inhibits platelet COX-1
without modifying the activity of systemic COX-1 or COX-2 . Because their
effects are reversible within the typical dosing interval, nonselective NSAIDs
(eg, ibu-profen) do not reproduce this effect, although naproxen, because of
its variably prolonged half-life, may provide antiplatelet benefit in some
individuals. TXA2, in addition to activating platelets, amplifies
the response to other platelet agonists; hence inhibition of its synthesis
inhibits secondary aggregation of platelets induced by adenosine diphosphate,
by low concentrations of thrombin and collagen, and by epinephrine. Not
surprisingly, selective COX-2 inhibitors do not alter platelet TXA2
biosynthesis and are not platelet inhibitors. However, COX-2-derived PGI2
generation is substantially suppressed during selective COX-2 inhibition,
removing a restraint on the cardiovascular action of TXA2, and other
platelet agonists. It is highly likely that selective depression of PGI2
generation contributes to the increased thrombotic events in humans treated
with selective COX-2 inhibitors.
Overview
analyses have shown that low-dose aspirin reduces the secondary incidence of
heart attack and stroke by about 25%. However, low-dose aspirin also elevates
the low risk of serious gastrointestinal bleeding about twofold over placebo.
Low-dose aspirin also reduces the incidence of first myocardial infarction.
However, in this case, the benefit versus risk of gastrointestinal bleeding is
less clear.
Respiratory System
PGE2
is a powerful bronchodilator when given in aerosol form. Unfortunately, it also
promotes coughing, and an analog that pos-sesses only the bronchodilator
properties has been difficult to obtain.
PGF2α and TXA2
are both strong bronchoconstrictors and were once thought to be primary
mediators in asthma. Polymorphisms in the genes for PGD2 synthase,
both DP receptors, and the TP recep-tor have been linked with asthma in humans.
DP antagonists, par-ticularly those directed against DP2, are being
investigated as potential treatments for allergic diseases including asthma.
However, the cysteinyl leukotrienes—LTC4, LTD4, and LTE4—probably
dominate during asthmatic constriction of the airways. As described, leukotriene-receptor inhibitors (eg, zafirlukast,montelukast) are effective
in asthma. A lipoxygenase inhibitor(zileuton)
has also been used in asthma but is not as popular as the receptor inhibitors.
It remains unclear whether leukotrienes are partially responsible for acute
respiratory distress syndrome.
Corticosteroids
and cromolyn are also useful in asthma. Corticosteroids inhibit eicosanoid
synthesis and thus limit the amounts of eicosanoid mediator available for
release. Cromolyn appears to inhibit the release of eicosanoids and other
mediators such as histamine and platelet-activating factor from mast cells.
Gastrointestinal System
The
word “cytoprotection” was coined to signify the remarkable protective effect of
the E prostaglandins against peptic ulcers inanimals at doses that do not
reduce acid secretion. Since then, numerous experimental and clinical
investigations have shown that the PGE compounds and their analogs protect
against peptic ulcers produced by either steroids or NSAIDs. Misoprostol is an orally active
synthetic analog of PGE1. The FDA-approved indication is for
prevention of NSAID-induced peptic ulcers. The drug is administered at a dosage
of 200 mcg four times daily with food. This and other PGE analogs (eg,
enprostil) are cytoprotective at low doses and inhibit gastric acid secretion
at higher doses. Misoprostol use is low, probably because of its adverse
effects including abdom-inal discomfort and occasional diarrhea. Dose-dependent
bone pain and hyperostosis have been described in patients with liver disease
who were given long-term PGE treatment.
Selective
COX-2 inhibitors were developed in an effort to spare gastric COX-1 so that the
natural cytoprotection by locally syn-thesized PGE2 and PGI2
is undisturbed . However, this benefit is seen only with highly selective
inhibitors and may be offset by increased cardiovascular toxicity.
Immune System
Cells
of the immune system contribute substantially to eico-sanoid biosynthesis
during an immune reaction. T and B lym-phocytes are not primary synthetic
sources; however, they may supply arachidonic acid to monocyte-macrophages for
eicosanoid synthesis. In addition, there is evidence for eicosanoid-mediated
cell-cell interaction by platelets, erythrocytes, leukocytes, and endothelial
cells.
The
eicosanoids modulate the effects of the immune system. PGE2 and PGI2
limit T-lymphocyte proliferation in vitro, as do corticosteroids. PGE2
also inhibits B-lymphocyte differentiation and the antigen-presenting function
of myeloid-derived cells, sup-pressing the immune response. T-cell clonal
expansion is attenu-ated through inhibition of interleukin-1 and interleukin-2
and class II antigen expression by macrophages or other antigen-presenting
cells. The leukotrienes, TXA2, and platelet-activating factor
stimu-late T-cell clonal expansion. These compounds stimulate the for-mation of
interleukin-1 and interleukin-2 as well as the expression of interleukin-2
receptors. The leukotrienes also promote interferon-γ release and can replace interleukin-2 as a
stimulator of interferon-γ. PGD2 induces chemotaxis and
migration of TH2 lymphocytes. These in vitro effects of the eicosanoids agree
with in vivo findings in animals with acute organ transplant rejection, as
described below.
A. Cell-Mediated Organ Transplant Rejection
Acute
organ transplant rejection is a cell-mediated immune response . Administration
of PGI2 to renal trans-plant patients has reversed the rejection
process in some cases. Experimental in vitro and in vivo data show that PGE2
and PGI2 can attenuate T-cell proliferation and rejection, which can
also be seen with drugs that inhibit TXA2 and leukotriene formation.
In organ transplant patients, urinary excretion of metabolites of TXA2
increases during acute rejection. Corticosteroids, the first-line drugs used
for treatment of acute rejection because of their lymphotoxic effects, inhibit
both phospholipase and COX-2 activity.
B. Inflammation
Aspirin
has been used to treat arthritis of all types for approxi-mately 100 years, but
its mechanism of action—inhibition of COX activity—was not discovered until
1971. COX-2 appears to be the form of the enzyme most associated with cells
involved in the inflammatory process, although, as outlined above, COX-1 also
contributes significantly to prostaglandin biosynthesis during inflammation.
C. Rheumatoid Arthritis
In
rheumatoid arthritis, immune complexes are deposited in the affected joints,
causing an inflammatory response that is amplified by eicosanoids. Lymphocytes
and macrophages accumulate in the synovium, whereas leukocytes localize mainly
in the synovial fluid. The major eicosanoids produced by leukocytes are
leukotrienes, which facilitate T-cell proliferation and act as
chemoattractants. Human macrophages synthesize the COX products PGE2
and TXA2 and large amounts of leukotrienes.
D. Infection
The
relationship of eicosanoids to infection is not well defined. The association
between the use of the anti-inflammatory steroids and increased risk of
infection is well established. However, NSAIDs do not seem to alter patient
responses to infection.
Glaucoma
Latanoprost,a stable long-acting PGF2αderivative, was the firstprostanoid used for glaucoma. The success of latanoprost has stimulated development of similar prostanoids with ocular hypotensive effects, and bimatoprost,travoprost, and unoprostoneare now available. These drugs act at the FP receptor and are administered as drops into the conjunctival sac once or twice daily. Adverse effects include irreversible brown pigmentation of the iris and eyelashes, drying of the eyes, and conjunctivitis.Related Topics