Chapter: Medicine and surgery: Nutritional and metabolic disorders
Porphyria - Metabolic disorders
Porphyria
Definition
The porphyrias are genetic or acquired deficiencies in the activity of enzymes in the heme biosynthetic pathway.
Aetiology/pathophysiology
Heme is synthesised from succinyl Co A and glycine (see Fig 13.2); differing enzyme deficiencies cause different patterns of disease.
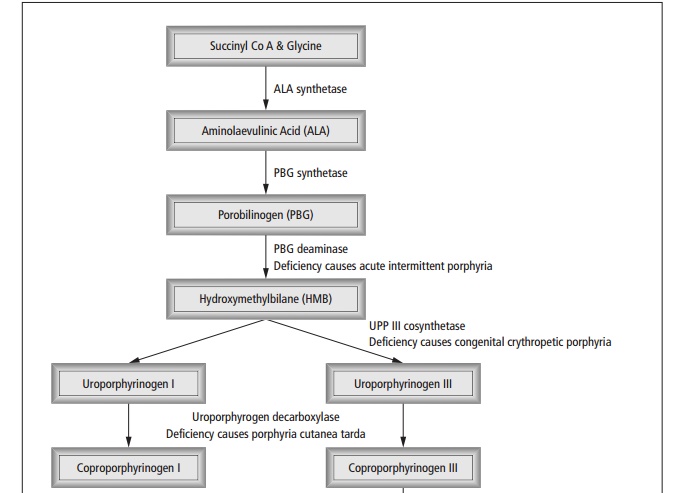
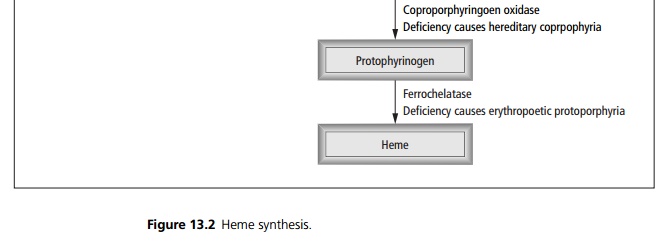
The first enzyme, ALA synthetase is the ratelimiting step. Enzyme deficiencies result in increases in metabolic intermediates, which are excreted and accumulate in tissues. This results in neurological, visceral symptoms and photocutaneous manifestations. Pophyrias may manifest as acute or chronic disease, the excess porphyrins deposit either in the liver (hepatic porphyrias) or in the bone marrow (erythropoetic porphyrias).
Clinical features/management
Acute intermittent porphyria is inherited in an autosomal dominant fashion. It presents in adult life with abdominal pain, vomiting and constipation, polyneuropathy, hypertension and tachycardia. Acute episodes are precipitated by alcohol and drugs. Diagnosis is suggested by leaving the urine to stand, which turns red brown.
Porphyria cutanea tarda presents with a bullous photodermatitis precipitated by alcohol. Urinary levels of uroporphyrinogen (the substrate for the deficient enzyme) are raised. Remission is induced by venesection.
Congenital erythropoietic porphyria is inherited in an autosomal recessive fashion. It causes an extreme photosensitivity with dystrophy and scarring.
Erythropoietic protoporphyria is inherited in an autosomal dominant fashion. The photosensitivity that results can be controlled with β -carotene by an un-known mechanism.
Related Topics