Chapter: Health Management in Aquaculture: Immunological and molecular biology techniques in disease diagnosis
Polymerase Chain Reaction (PCR) - Aquaculture Molecular biology techniques in disease diagnosis
Polymerase Chain Reaction (PCR)
Various molecular biology techniques continue becoming more important infish and srimp farming, particularly in detection and prevention of various diseases. One of the most prominent techniques is the PCR. No technique has had a greater impact on the practice of molecular biology than the PCR. The PCR for amplification of specific nucleic acid sequences was introduced by Saiki et al. (1985) and has subsequently proved to be one of the most important scientific innovations of the past decade. With this technique, one can rapidly detect a virus or bacteria, few copies of mRNA, rapidly synthesize, clone and sequence virtually any segment of DNA. Despite the incredible power of the technique there has been one major limitation that is the DNA must be ex-tracted from the sample one can not correlate PCR results with the pathological features of material being tested.
The development of PCR means that small amounts of DNA no longer limit molecular biology research or DNA-based diagnostic procedures. The tech-nique is continuously improving and its full impact on molecular diagnostics is yet to come. PCR currently has many applications, including analysis of an-cient DNA from fossils, amplification of small DNA amounts for analysis by DNA fingerprinting, mapping the human genome and also those of other spe-cies, and detection of microorganisms present in low densities in water, food, soil or other organisms. In aquaculture, PCR is a valuable tool for the prevention, control and management of various dis-eases.
For fish and shrimp farmers, it permits fast, widespread, and sensitive screening of virus carriers, and also for early or light infections. The tests can be carried out non-destructively by us-ing body fragments, blood or feces from fish and shrimp tested. PCR can be used to screen both broodstock animals and also larvae before stock-ing. PCR is rapidly becoming a critical instrument to detect fish and shrimp pathogens.
PCR assay has been widely used in the detection of fish viruses like stripe jack nervous necrosis vi-rus (SJNNV) (Nishizawa et al., 1994), red sea bream iridovirus (RSIV) (Kurita et al., 1998), aquatic birnaviruses (Williams et al., 1999) and shrimp viruses like white spot syndrome virus (WSSV), monodon baculovirus (MBV), infectious hypodermal and hematopoietic necrosis virus (IHHNV), hepatopancreatic parvo virus (HPV), baculovirus penaei (BP), Taura syndrome virus (TSV), yellow head virus (YHV) and baculoviral midgut gland necrosis virus (BMN) (Lightner, 1996; Wongteerasupaya et al., 1997; Lightner and Redman, 1998; Tapay et al., 1999; Hsu et al., 2000; Magbanua et al., 2000). The assay has also been used in the detection of Vibrio penaeicida in shrimp (Genmoto et al., 1996; Nakai et al., 1997)
Aeromonas salmonicidasubspecies salmonicida (Miyata et al., 1996), Pasteurella piscicida (Aoki et al., 1997) and Lactococcus garvieae (Aoki et al., 2000) in fish. PCR uses a thermostable polymerase to produce multiple copies of specific nucleic acid region quickly and exponentially. For example, starting with a single copy of a 1 kb DNA sequence, 1011 copies (or 100 ng) of the same sequence can be produced within a few hours. Once the reaction has occurred, a number of methods for identifica-tion and characterization of the amplification products are then applicable, of which the sim-plest is to identify the products according to their size following migration in the agarose gels. For many diagnostic applications, the simple visual-ization of a PCR product of characteristic size is a significant outcome since it indicates the presence of the target DNA sequence in the original sample.
Basic principle
Each PCR amplification is subdivided into three steps which are repeated in cycles as indicated below (Figure 10-12):
(1) melting or denaturation (strand separation) of the double strand DNA(one to several minutes at 94-96°C);
(2) annealing of the two primers to opposite DNA strands (one to severalminutes at 50-65°C); and
(3) extensionof the primers by polymerase-mediated nucleotide additionsto produce two copies of the original sequence (one to several minutes at 72°C).
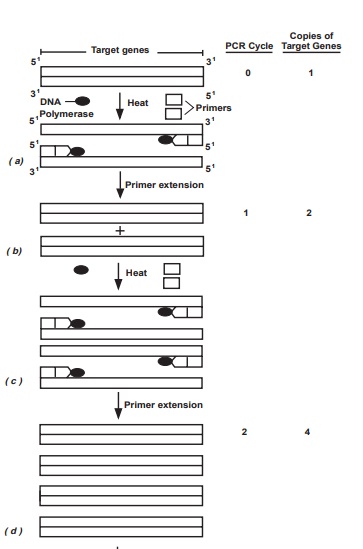
During cellular DNA replication, enzymes first unwind and denature the DNA double helix into single strands. After the DNA is denatured, one more event must occur before DNA synthesis may be catalyzed by the DNA polymerase. It must find an area of transition from single stranded to double stranded DNA. At proper temperature typically around 55°C, sufficient primer-target DNA hy-bridization occurs which leads to the synthesis of complementary strands es-sential for the amplification step. During PCR, high temperature is used to separate the DNA molecules into single strands, and synthetic sequences of single-stranded DNA (20-30 nucleotides) serve as primers. Two different primer sequences are used to bracket the target region to be amplified. One primer is complementary to one DNA strand at the beginning of the target region; a second primer is complementary to a sequence on the opposite DNA strand at the end of the target region.
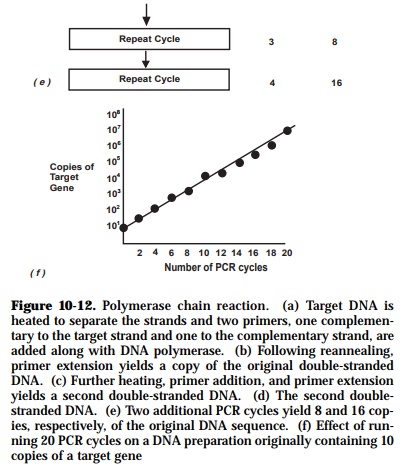
As amplification proceeds, the DNA sequence between the primers doubles after each cycle. Following thirty cycles, a theoritical amplification factor of one billion is attained.assuming 100% efficiency during each cycle. The final number of copies of the target sequence is expressed by the formula, (2n - 2n)x, where:
n - number of cycles;
2n - first product obtained after cycle 1 and second products obtained after cycle 2 with undefined length;
x - number of copies of the original template.
There are factors that act against the process being 100% efficient at each cycle. Their effect is more pronounced in the later cycles of PCR. Normally, the amount of enzyme becomes limiting after 25-30 cycles, which corresponds to about 106-fold amplification, due to molar target excess. The enzyme activity also becomes limiting due to thermal denaturation of the enzyme during the process. Another factor is the reannealing of target strands as their concentra-tion increases. The reannealing of target strands then competes with primer annealing.
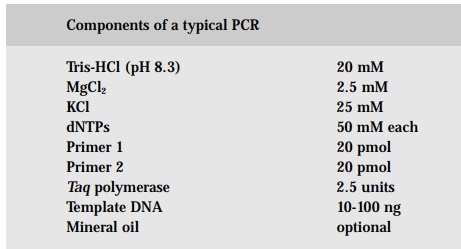
Two important innovations were responsible for automating PCR. First, a heat-stable DNA polymerase was isolated from the bacterium Thermus aquaticus that lives in hot springs. Hence, the term Taq (DNA) polymerase came to be. This enzyme remains active despite repeated heating during many cycles of amplification. Second, thermal cyclers were invented which automatically control the repetitive temperature changes required for PCR.
Following amplification, the PCR products are usually loaded into wells of an agarose gel and electrophoresed. Since PCR amplifications can generate micro-gram quantities of product, amplified fragments can be visualized easily fol-lowing staining with a chemical stain such as ethidium bromide. The important point to remember is that the amplification is selective - only the DNA sequence located between the primers is amplified exponentially. The rest of the DNA in the genome is not amplified and remains invisible in the gel.
Primer design
Some simple rules aid in the design of efficient primers:
1. Typical primers are 18 to 28 nucleotides in length having 50 to 60% G+C composition. 2. The calculated melting temperatures for a given primer pair should be balanced. One can use the rule-of-thumb calculation of 2°C for A or T and 4°C for G or C. Depending on the application, melting tem- peratures between 55°C and 80°C are desired.
2. To prevent self-annealing, primers should not be complementary. This pre- caution is critical at the extreme 3’ ends where any complementarity may lead to considerable primer-dimer formation and reduces the yield of the desired product.
3. Runs (three or more) of C’s or G’s at the 3’ ends of primers may promote mispriming at G+C-rich sequences and should be avoided when possible.
If all else fail, it usually helps to try a different primer pair. A less obvious reason for some primers failing to work is the presence of secondary structure in the template DNA. Software is also available from many commercial and academic sources to assist in the process. Most software packages for DNA sequence analysis now include menus for PCR primer design.
Nested PCR
Nested PCR primers are ones that are internal to the first primer pair. The larger fragment produced by the first round of PCR is used as the template for the second PCR. Nested PCR can also be performed with one of the first primer pair and a single nested primer. The sensitivity and specificity of both DNA and RNA amplification can be dramatically increased by using this method. The specificity is particularly enhanced because this technique almost always eliminates any spurious nonspecific amplification products. This is becauseafter the first round of PCR, any nonspecific products are unlikely to be suffi-ciently complementary to the nested primers to be able to serve as a template for further amplification, thus the desired target sequence is preferentially am-plified. However, the increased risk of contamination is a drawback of this extreme sensitivity.
RNA (RT) PCR
PCR amplifies DNA sequences. In order to perform PCR on RNA sequences using Taq DNA polymerase, the RNA must first be transcribed into a cDNA (complementary DNA) copy of the RNA sequence because Taq has limited re-verse transcriptase activity. This is called reverse transcription (RT). Thus, RNA amplification is achieved by the reverse transcription-polymerase chain reaction (RT-PCR). There are several different kinds of primers that can be used to make cDNAs, like oligo-dT will prime cDNA synthesis on all polyadenylated RNAs, random-primed cDNA synthesis gives a broad range of cDNAs and is not limited to polyadenylated RNAs and lastly, oligo-nucleotide primers complementary to the RNA(s) of interest may be used to synthesize highly specific cDNAs.
One-tube RT-PCR incorporates both the reverse transcriptase enzyme and a thermostable DNA polymerase in a single tube for synthesis and amplification of the target RNA sequence. This is the preferred procedure for routine analy-sis. Commercial RT-PCR kits are available and alternatively, reagent mixes can be prepared also from separate component parts.
Problems with PCR
PCR is an extremely powerful technique, but its very power can also lead to considerable problems, particularly when detecting virus or bacterial genes for diagnostic purposes. It is important to remember that nucleic acid from dead as well as viable microorganisms will give a positive reaction. Since even a single molecule of DNA can be amplified by PCR, it is also vital to prevent cross-contamination of DNA samples with amplified or foreign DNA. The slightest contamination of glassware, pipettes or reagents can result in the pro-duction of false-positive reactions. Such contamination problems impose a need for extreme cleanliness and rigorous controls. Amplification reactions should be performed in physical isolation (i.e. in a different room) from the parts of the laboratory where specimens are received and target nucleic acid is prepared. Various techniques for reducing extraneous DNA contamination of PCR products have been described, but it is vital that each set of PCR amplifi-cations should include control reactions to verify the purity of reagents and the cleanliness of equipment.
PCR is vulnerable to contamination that will cause erroneous results. False positives will result from contamination of the reaction with target RNA or RT-PCR products. False negatives can be caused by the presence of inhibitors in the test samples or badly degraded target materials. For competent PCR, the sample must be either fresh and in good condition prior to nucleic acid extrac-tion or preserved to maintain nucleic acid suitable for extraction. Proper con-sideration to the extraction procedure is also important. Maintenance of rigor-ously clean experimental techniques, use of standard reaction conditions and inclusion of internal standards as positive and negative controls are essential to gain accurate interpretation of the results.
Other problems may arise from the relatively high error rate of Taq polymerase. Base substitutions occur at about one in every 9,000 bp, and frameshifts at about one in every 40,000 bp. Although such error rates may seem to be insig-nificant, they may have profound effects on the homogeneity of the amplified products.
Although PCR can now be semi-automated because of the availability of the thermocycler, the technique still requires a certain amount of technical skill and some specialized equipments to prepare samples and perform amplifica-tion reactions successfully.
Related Topics