Chapter: Clinical Anesthesiology: Anesthetic Management: Neurophysiology & Anesthesia
Physiology of Brain Protection
Physiology of Brain Protection
PATHOPHYSIOLOGY OF CEREBRAL ISCHEMIA
The
brain is very vulnerable to ischemic injury because of its relatively high
oxygenconsumption and near total dependence on aero-bic glucose metabolism
(above). Interruption of cerebral perfusion, metabolic substrate (glucose), or
severe hypoxemia rapidly results in functional impairment; reduced perfusion
also impairs clear-ance of potentially toxic metabolites. If normal oxygen
tension, blood flow, and glucose supply are not reestablished within 3–8 min
under most conditions, ATP stores are depleted, and irrevers-ible neuronal
injury begins. When CBF decreases below 10 mL/100 g/min, cell function is
deranged, and ion pumps fail to maintain cellular vitality. The ratio of
lactate to pyruvate is increased second-ary to anaerobic metabolism. During
ischemia, intracellular K +
decreases, and intracellular Na +
increases. More importantly, intracellular Ca 2+
increases because of failure of ATP-dependent pumps to either extrude the ion
extracellularly or into intracellular cisterns, increased intracellular Na+
concentration, and release of the excitatory neurotransmitter glutamate.
Glutamate acts at the NMDA receptor, further enhancing Ca2+
entry into the cell, hence the potential benefit of NMDA blockers for
neuroprotection.
Sustained
increases in intracellular Ca 2+ activate lipases and proteases,
which initiate and propagate structural damage to neurons. Increases in free
fatty acid concentration and cyclooxygenase and lipoxy-genase activities result
in the formation of prosta-glandins and leukotrienes, some of which are potent
mediators of cellular injury. Accumulation of toxic metabolites, such as lactic
acid, also impairs cellu-lar function and interferes with repair mechanisms.
Lastly, reperfusion of ischemic tissues can cause additional tissue damage due
to the formation of oxygen-derived free radicals. Likewise, inflamma-tion and
edema can promote further neuronal dam-age, leading to cellular apoptosis.
STRATEGIES FOR BRAIN PROTECTION
Ischemic brain injury is usually classified as focal
(incomplete) or global (complete). Global ischemia includes total circulatory
arrest as well as global hypoxia. Cessation of perfusion may be caused by
cardiac arrest or deliberate circulatory arrest, whereas global hypoxia may be
caused by severe respiratory failure, drowning, and asphyxia (includ-ing
anesthetic mishaps). Focal ischemia includes embolic, hemorrhagic, and
atherosclerotic strokes, as well as blunt, penetrating, and surgical trauma.
In some instances, interventions aimed at restoring perfusion
and oxygenation are possible; these include reestablishing effective
circulation, normalizing arterial oxygenation and oxygen-car-rying capacity, or
reopening an occluded vessel. With focal ischemia, the brain tissue surrounding
a severely damaged area may suffer marked func-tional impairment but still
remain viable. Such areas are thought to have very marginal perfusion (<15 mL/100 g/min), but, if further injury can be lim-ited and
normal flow is rapidly restored, these areas (the “ischemic penumbra”) may
recover completely. When the above interventions are not applicable or
available, the emphasis must be on limiting the extent of brain injury.
From a practical point of view, efforts
aimed at preventing or limiting neuronal tissue damage are often the same
whether the ischemia is focal or global. Clinical goals are usually to optimize
CPP, decrease metabolic requirements (basal and electri-cal), and possibly
block mediators of cellular injury. Clearly, the most effective strategy is
prevention, because once injury has occurred, measures aimed at cerebral
protection become less effective.
Hypothermia
Hypothermia is an eff ective method for
pro-tecting the brain during focal and global isch-emia. Indeed, profound
hypothermia is often used for up to 1 hr of total circulatory arrest. Unlike
anesthetic agents, hypothermia decreases both basal and elec-trical metabolic
requirements throughout the brain; metabolic requirements continue to decrease
even after complete electrical silence. Additionally, hypo-thermia reduces free
radicals and other mediators of ischemic injury. Induced hypothermia has shown
benefit following cardiac arrest and is a routine part of most postarrest
protocols for comatose patients.
Anesthetic Agents
Barbiturates, etomidate, propofol, and
isoflurane can produce complete electrical silence of the brain and eliminate
the metabolic cost of electrical activity; unfortunately, these agents have no
effect on basal energy requirements. Furthermore, with the excep-tion of
barbiturates, their effects are nonuniform,
affecting different parts of the brain to variable extents.
Ketamine may also have a protective effect because of its
ability to block the actions of gluta-mate at the NMDA) receptor.
No anesthetic agent has consistently been shown to be protective
against global ischemia. The ever increasing number of studies highlighting the
potential neurotoxicity of anesthetics (especially in infants) also questions
the role of volatile anesthetics in neuroprotection.
Specific Adjuncts
Nimodipine plays a role in the in the treatment of vasospasm
associated with subarachnoid hemor-rhage. Studies are ongoing to discern the
roles of various NMDA receptor antagonists, erythropoi-etin, Ca2+ antagonists, and free
radical scavengers to mitigate ischemic neuronal injury.
General Measures
Maintenance of a satisfactory CPP is
critical. Thus, arterial blood pressure should be normal or slightly elevated,
and increases in venous and ICP should be avoided. Oxygen-carrying capacity
should be maintained and normal arterial oxygen tension preserved. Hyperglycemia
aggravates neurological injuries following either focal or global ischemia, and
blood glucose should be maintained at less than 180 mg/dL. Normocarbia should
be maintained, as both hypercarbia and hypocarbia have no benefi-cial effect in
the setting of ischemia and could prove detrimental; hypocarbia-induced
cerebral vaso-constriction may aggravate the ischemia, whereas hypercarbia may
induce a steal phenomenon (with focal ischemia) or worsen intracellular
acidosis.
EFFECT OF ANESTHESIA ON ELECTROPHYSIOLOGICAL MONITORING
Electrophysiological monitors are used
to assess the functional integrity of the CNS. The most com-monly used monitor
for neurosurgical procedures is evoked potentials. EEG is much less commonly
used. Proper application of these monitoring modalities
is critically dependent on monitoring the specific area at risk
and recognizing anesthetic-induced changes.
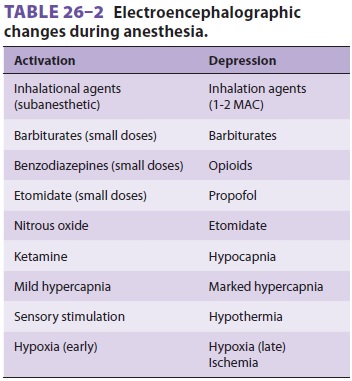
The effects of anesthetic agents on the EEG are summarized in Table26–2.
ELECTROENCEPHALOGRAPHY
EEG monitoring is useful for assessing
the adequacy of cerebral perfusion during carotid endarterectomy (CEA), as well
as anesthetic depth (most often with processed EEG). EEG changes can be
simplistically described as either activation or depression. EEG activation (a
shift to predominantly high-frequency and low-voltage activity) is seen with
light anesthe-sia and surgical stimulation, whereas EEG depres-sion (a shift to
predominantly low-frequency and high-voltage activity) occurs with deep anesthesia
or cerebral compromise. Most anesthetics produce an EEG consisting of an
initial activation (at sub-anesthetic doses) followed by dose-dependent
depression.
Inhalation Anesthetics
Isoflurane can produce an isoelectric
EEG at high clinical doses (1–2 MAC). Desflurane and sevoflurane produce a
burst suppression pattern at high doses (>1.2 and >1.5 MAC, respectively)
but not electrical silence. Nitrous oxide is alsounusual
in that it increases both frequency and amplitude (high-amplitude activation).
Intravenous Agents
Benzodiazepines can produce both activation and depression of
the EEG. Barbiturates, etomidate, and propofol produce a similar pattern and
are the only intravenous agents capable of producing burst sup-pression and
electrical silence at high doses. In contrast, opioids characteristically
produce only dose-dependent depression of the EEG. Lastly, ket-amine produces
an unusual activation consisting of rhythmic high-amplitude theta activity
followed by very high-amplitude gamma and low-amplitude beta activities.
EVOKED POTENTIALS
Somatosensory evoked potentials test the integrity of the spinal
dorsal columns and the sensory cortex and may be useful during resection of
spinal tumors, instrumentation of the spine, CEA, and aortic surgery. The adequacy
of perfusion of the spinal cord during aortic surgery is probably better
assessed with motor evoked potentials (which assess the anterior part of the
spinal cord). Brainstem auditory evoked potentials test the integrity of the
eighth cranial nerve and the audi-tory pathways above the pons and are used for
surgery in the posterior fossa. Visual evoked potentials may be used to monitor
the optic nerve and occipital cortex during resections of large pituitary
tumors.
Interpretation of evoked potentials is more
complicated than that of the EEG. Evoked potentials have poststimulus latencies
that are described as short, intermediate, and long. Short-latency evoked
potentials arise from the nerve stimulated or the brain stem. Intermediate- and
long-latency evoked potentials are primarily of cortical origin. In general,
short-latency potentials are least affected by anes-thetic agents, whereas
long-latency potentials are affected by even subanesthetic levels of most
agents. Visual evoked potentials are most affected by anes-thetics, whereas
brain stem auditory evoked poten-tials are least affected.Intravenous agents in
clinical doses gener-ally have less marked effects on evoked potentials than do
volatile agents, but, in high doses, can also decrease amplitude and increase
latencies.
Related Topics