Chapter: Basic & Clinical Pharmacology : Sedative-Hypnotic Drugs
Pharmacokinetics - Basic Pharmacology of Sedative Hypnotics
Pharmacokinetics
A. Absorption and Distribution
The
rates of oral absorption of sedative-hypnotics differ depending on a number of
factors, including lipophilicity. For example, the absorption of triazolam is
extremely rapid, and that of diazepam and the active metabolite of clorazepate
is more rapid than other commonly used benzodiazepines. Clorazepate, a prodrug,
is converted to its active form, desmethyldiazepam (nordiazepam), by acid
hydrolysis in the stomach. Most of the barbiturates and other older
sedative-hypnotics, as well as the newer hypnotics (eszopiclone, zaleplon,
zolpidem), are absorbed rapidly into the blood following oral administration.
Lipid
solubility plays a major role in determining the rate at which a particular
sedative-hypnotic enters the central nervous system. This property is
responsible for the rapid onset of central nervous system effects of triazolam,
thiopental , and the newer hypnotics eszopiclone, zaleplon, and zolpidem.
All
sedative-hypnotics cross the placental barrier during pregnancy. If
sedative-hypnotics are given during the predelivery period, they may contribute
to the depression of neonatal vital functions. Sedative-hypnotics are also
detectable in breast milk and may exert depressant effects in the nursing
infant.
B. Biotransformation
Metabolic
transformation to more water-soluble metabolites is necessary for clearance of
sedative-hypnotics from the body. The microsomal drug-metabolizing enzyme
systems of the liver are most important in this regard, so elimination
half-life of these drugs depends mainly on the rate of their metabolic
transformation.
1. Benzodiazepines—Hepatic metabolism
accounts for theclearance of all benzodiazepines. The patterns and rates of
metab-olism depend on the individual drugs. Most benzodiazepines undergo
microsomal oxidation (phase I reactions), including N-dealkylation and aliphatic hydroxylation catalyzed by cyto-chrome
P450 isozymes, especially CYP3A4. The metabolites are subsequently conjugated
(phase II reactions) to form glucuronides that are excreted in the urine.
However, many phase I metabolites of benzodiazepines are pharmacologically
active, some with long half-lives (Figure 22–5). For example,
desmethyldiazepam, which has an elimination half-life of more than 40 hours, is
an activemetabolite of chlordiazepoxide, diazepam, prazepam, and clorazepate.
Alprazolam and triazolam undergo α-hydroxylation, and the resulting metabolites
appear to exert short-lived pharma-cologic effects because they are rapidly
conjugated to form inactive glucuronides. The short elimination half-life of
triazolam (2–3 hours) favors its use as a hypnotic rather than as a sedative
drug.
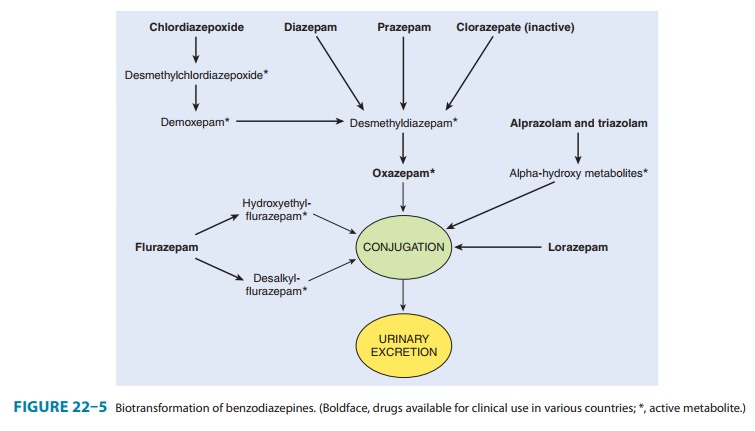
The
formation of active metabolites has complicated studies on the pharmacokinetics
of the benzodiazepines in humans because he elimination half-life of the parent
drug may have little relation to the time course of pharmacologic effects.
Benzodiazepines for which the parent drug or active metabolites have long
half-lives are predictably more likely to cause cumulative effects with
mul-tiple doses. Cumulative and residual effects such as excessive drowsiness
appear to be less of a problem with such drugs as estazolam, oxazepam, and
lorazepam, which have relatively short half-lives and are metabolized directly
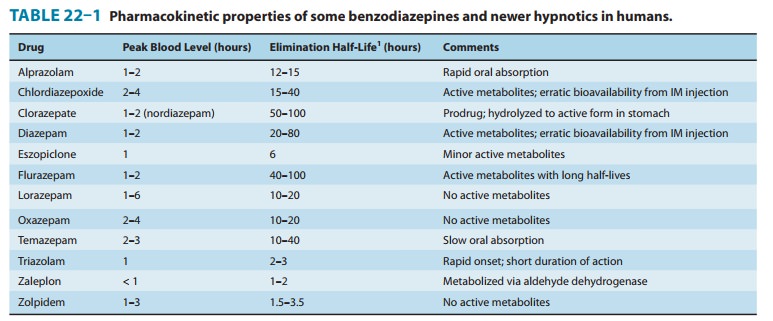
to inactive glucuronides. Some pharmacokinetic properties of selected
benzodiazepines are listed in Table 22–1. The metabolism of several commonly
used benzodiazepines including diazepam, midazolam, and triazolam is affected
by inhibitors and inducers of hepatic P450 isozymes .
Ramelteon
Melatonin receptors are thought to be involved in maintain-ing circadian rhythms underlying the sleep-wake cycle . Ramelteon (Rozerem), a novel hypnotic drug specifically useful for patients who have difficulty in falling asleep, is an agonist at MT1 and MT2 melatonin receptors located in the suprachiasmatic nuclei of the brain. The drug has no direct effects on GABAergic neurotransmission in the central nervous system. In polysomnography studies of patients with chronic insomnia, ramelteon reduced the latency of persistent sleep with no effects on sleep architec-ture and no rebound insomnia or significant withdrawal symptoms. Ramelteon has minimal potential for abuse, is not a controlled substance, and regular use does not result in dependence. The drug is rapidly absorbed after oral adminis-tration and undergoes extensive first-pass metabolism, form-ing an active metabolite with longer half-life (2–5 hours) than the parent drug. The CYP1A2 isoform of cytochrome P450 is mainly responsible for the metabolism of ramelteon, but the CYP2C9 isoform is also involved. The drug should not be used in combination with inhibitors of CYP1A2 (eg, ciprofloxacin, fluvoxamine, tacrine, zileuton) or CYP2C9 (eg, fluconazole) and should be used with caution in patients with liver dys-function. The CYP inducer rifampin markedly reduces the plasma levels of both ramelteon and its active metabolite. Adverse effects of ramelteon include dizziness, somnolence, fatigue, and endocrine changes as well as decreases in testos-terone and increases in prolactin. Ramelteon is an FDA preg-nancy category C drug.
Barbiturates—With the exception of
phenobarbital, onlyinsignificant quantities of the barbiturates are excreted
unchanged. The major metabolic pathways involve oxidation by hepatic enzymes to
form alcohols, acids, and ketones, which appear in theurine as glucuronide
conjugates. The overall rate of hepatic metabolism in humans depends on the
individual drug but (with the exception of the thiobarbiturates) is usually
slow. The elimina-tion half-lives of secobarbital and pentobarbital range from
18 to 48 hours in different individuals. The elimination half-life of
phe-nobarbital in humans is 4–5 days. Multiple dosing with these agents can
lead to cumulative effects.
Newer hypnotics—After oral
administration of the standardformulation, zolpidem reaches peak plasma levels
in 1.6 hours. A biphasic release formulation extends plasma levels by
approximately 2 hours. Zolpidem is rapidly metabolized to inactive metabolites
via oxidation and hydroxylation by hepatic cytochromes P450 including the
CYP3A4 isozyme. The elimination half-life of the drug is 1.5– 3.5 hours, with
clearance decreased in elderly patients. Zaleplon is metabolized to inactive
metabolites, mainly by hepatic aldehyde oxidase and partly by the cytochrome
P450 isoform CYP3A4. The half-life of the drug is about 1 hour. Dosage should
be reduced in patients with hepatic impairment and in the elderly. Cimetidine,
which inhibits both aldehyde dehydrogenase and CYP3A4, mark-edly increases the
peak plasma level of zaleplon. Eszopiclone is metabolized by hepatic
cytochromes P450 (especially CYP3A4) to form the inactive N-oxide derivative and weakly active desmethyl-eszopiclone. The
elimination half-life of eszopiclone is approxi-mately 6 hours and is prolonged
in the elderly and in the presence of inhibitors of CYP3A4 (eg, ketoconazole).
Inducers of CYP3A4 (eg, rifampin) increase the hepatic metabolism of
eszopiclone.
Buspirone
Buspirone has selective anxiolytic effects, and its pharmaco-logic
characteristics differ from those of other drugs. Buspirone relieves anxiety
without causing marked sedative, hypnotic, or euphoric effects. Unlike
benzo-diazepines, the drug has no anticonvulsant or muscle relaxant properties.
Buspirone does not interact directly with GABAergic systems. It may exert its
anxiolytic effects by act-ing as a partial agonist at brain 5-HT1A
receptors, but it also has affinity for brain dopamine D 2
receptors. Buspirone-treated patients show no rebound anxiety or withdrawal
signs on abrupt discontinuance. The drug is not effective in blocking the acute
withdrawal syndrome resulting from abrupt cessation of use of benzodiazepines
or other sedative-hypnotics. Buspirone has minimal abuse liability. In marked
contrast to the benzodiazepines, the anxiolytic effects of bus-pirone may take
more than a week to become established, making the drug unsuitable for
management of acute anxiety states. The drug is used in generalized anxiety
states but is less effective in panic disorders.
Buspirone is rapidly absorbed orally but undergoes exten-sive first-pass
metabolism via hydroxylation and dealkylation reactions to form several active
metabolites. The major metabolite is 1-(2-pyrimidyl)-piperazine (1-PP), which
has α2-adrenoceptor–blocking actions and which enters the cen-tral
nervous system to reach higher levels than the parent drug. It is not known
what role (if any) 1-PP plays in the central actions of buspirone. The
elimination half-life of buspirone is 2–4 hours, and liver dysfunction may slow
its clearance. Rifampin, an inducer of cytochrome P450, decreases the half-life
of buspirone; inhibitors of CYP3A4 (eg, erythromycin, ketoconazole, grapefruit
juice, nefazodone) can markedly increase its plasma levels.
C. Excretion
The
water-soluble metabolites of sedative-hypnotics, mostly formed via the
conjugation of phase I metabolites, are excreted mainly via the kidney. In most
cases, changes in renal function do not have a marked effect on the elimination
of parent drugs. Phenobarbital is excreted unchanged in the urine to a certain
extent (20–30% in humans), and its elimination rate can be increased
significantly by alkalinization of the urine. This is partly due to increased
ionization at alkaline pH, since phenobarbital is a weak acid with a pKa
of 7.4.
D. Factors Affecting Biodisposition
The
biodisposition of sedative-hypnotics can be influenced by several factors,
particularly alterations in hepatic function result-ing from disease or
drug-induced increases or decreases in microsomal enzyme activities
In
very old patients and in patients with severe liver disease, the elimination
half-lives of these drugs are often increased signifi-cantly. In such cases,
multiple normal doses of these sedative-hypnotics can result in excessive
central nervous system effects.
The
activity of hepatic microsomal drug-metabolizing enzymes may be increased in
patients exposed to certain older sedative-hypnotics on a long-term basis
(enzyme induction;). Barbiturates (especially phenobarbital) and meprobamate
are most likely to cause this effect, which may result in an increase in their
own hepatic metabolism as well as that of other drugs. Increased
biotransformation of other pharmacologic agents as a result of enzyme induction
by barbiturates is a potential mechanism under-lying drug interactions . In
contrast, benzodiaz-epines and the newer hypnotics do not change hepatic
drug-metabolizing enzyme activity with continuous use.
Related Topics