Chapter: Basic & Clinical Pharmacology : Sedative-Hypnotic Drugs
Pharmacodynamics of Benzodiazepines, Barbiturates, & Newer Hypnotics
Pharmacodynamics of
Benzodiazepines, Barbiturates, & Newer Hypnotics
A. Molecular Pharmacology of the GABAA Receptor
The
benzodiazepines, the barbiturates, zolpidem, zaleplon, eszopi-clone, and many
other drugs bind to molecular components of the GABAA receptor in
neuronal membranes in the central nervous system. This receptor, which
functions as a chloride ion channel, is activated by the inhibitory
neurotransmitter GABA .
The
GABAA receptor has a pentameric structure assembled from five
subunits (each with four membrane-spanning domains) selected from multiple
polypeptide classes (α, β, γ, δ, ε, π, ρ, etc). Multiple subunits of several of these
classes have been character-ized, among them six different α (eg, α1 through α6), four β, and three γ. A model of the GABAA
receptor-chloride ion channel macromolecular complex is shown in Figure 22–6.
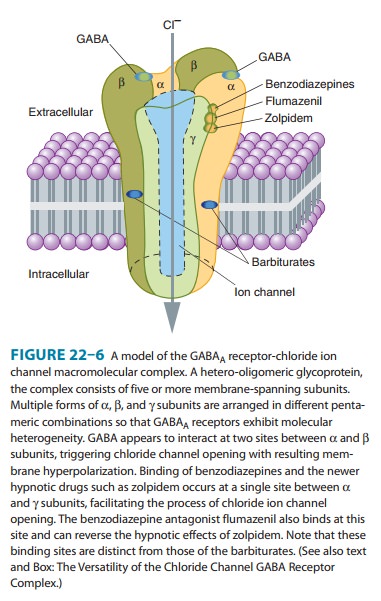
A major isoform of the GABAA receptor that is found in many regions of the brain consists of two α1, two β2, and one γ2 subunits. In this isoform, the two binding sites for GABA are located between adjacent α1 and β2 subunits, and the binding pocket for benzodiazepines (the BZ site of the GABAA receptor) is between anα1 and the γ2 subunit. However, GABAA receptors in different areas of the central nervous system consist of various combinations of the essential subunits, and the benzodiazepines bind to many of these, including receptor isoforms containing α2, α3, and α5 subunits.
Barbiturates also bind to multiple isoforms of
the GABAA receptor but at different sites from those with which
benzodiazepines interact. In contrast to benzodiazepines, zolpidem, zaleplon,
and eszopiclone bind more selectively because these drugs interact only with
GABAA-receptor isoforms that contain α1 subunits. The heterogeneity of GABAA
receptors may constitute the molecular basis for the varied pharmacologic
actions of benzodiazepines and related drugs (see Box: GABA Receptor
Heterogeneity & Pharmacologic Selectivity).
In
contrast to GABA itself, benzodiazepines and other sedative-hypnotics have a
low affinity for GABAB receptors, which are acti-vated by the
spasmolytic drug baclofen.
B. Neuropharmacology
GABA
(γ-aminobutyric
acid) is a major inhibitory neurotransmitter in the central nervous system .
Electrophysiologic studies have shown that benzodiazepines potentiate GABAergic
inhi-bition at all levels of the neuraxis, including the spinal cord,
hypo-thalamus, hippocampus, substantia nigra, cerebellar cortex, and cerebral
cortex. Benzodiazepines appear to increase the efficiency of GABAergic synaptic
inhibition. The benzodiazepines do not substi tute for GABA but appear to
enhance GABA’s effects allosterically without directly activating GABAA
receptors or opening the associ-ated chloride channels. The enhancement in
chloride ion conduc-tance induced by the interaction of benzodiazepines with
GABA takes the form of an increase in the frequency
of channel-opening events.
Barbiturates
also facilitate the actions of GABA at multiple sites in the central nervous
system, but—in contrast to benzodiazepines—they appear to increase the duration of the GABA-gated chloride
channel openings. At high concentrations, the barbiturates may also be
GABA-mimetic, directly activating chloride channels. These effects involve a
binding site or sites distinct from the benzodiazepine binding sites.
Barbiturates are less selective in their actions than benzodiazepines, because
they also depress the actions of the excitatory neurotransmitter glu-tamic acid
via binding to the AMPA receptor. Barbiturates also exert nonsynaptic membrane
effects in parallel with their effects on GABA and glutamate neurotransmission.
This multiplicity of sites of action of barbiturates may be the basis for their
ability to induce full surgical anesthesia
and for their more pronounced central depressant effects (which result
in their low margin of safety) compared with benzodiazepines and the newer
hypnotics.
C. Benzodiazepine Binding Site Ligands
The
components of the GABAA receptor-chloride ion channel mac-romolecule
that function as benzodiazepine binding sites exhibit heterogeneity (see Box:
The Versatility of the Chloride Channel GABA Receptor Complex). Three types of
ligand-benzodiazepine receptor interactions have been reported: (1) Agonists facilitate GABA actions, and
this occurs at multiple BZ binding sites in the case of the benzodiazepines. As
noted above, the nonbenzodiaz-epines zolpidem, zaleplon, and eszopiclone are
selective agonists at the BZ sites that contain anα1 subunit. Endogenous agonist ligands for the
BZ binding sites have been proposed, because benzodiazepine-like chemicals have
been isolated from brain tissue of animals never exposed to these drugs.
Nonbenzodiazepine molecules that have affinity for BZ sites on the GABAA
receptor have also been detected in human brain. (2) Antagonists are typi-fied by the synthetic benzodiazepine
derivative flumazenil, which blocks
the actions of benzodiazepines, eszopiclone, zaleplon, and zolpidem but does
not antagonize the actions of barbiturates, mep-robamate, or ethanol. Certain
endogenous neuropeptides are also capable of blocking the interaction of
benzodiazepines with BZ binding sites. (3) Inverse
agonists act as negative allosteric modula-tors of GABA-receptor function .
Their interaction with BZ sites on the GABAA receptor can produce anxiety and sei-zures, an action
that has been demonstrated for several compounds, especially the β-carbolines, eg, n-butyl-β-carboline-3-carboxylate (β-CCB). In addition to
their direct actions, these molecules can block the binding and the effects of
benzodiazepines.
The
physiologic significance of endogenous modulators of GABA functions in the
central nervous system remains unclear. To date, it has not been established
that the putative endogenous ligands of BZ binding sites play a role in the
control of states of anxiety, sleep patterns, or any other characteristic
behavioral expression of central nervous system function.
GABA Receptor Heterogeneity & Pharmacologic Selectivity
Studies involving strains of genetically engineered
(“knock-out”) rodents have demonstrated that the specific pharmaco-logic
actions elicited by benzodiazepines and other drugs that modulate GABA actions
are influenced by the composition of the subunits assembled to form the GABA A
receptor. Benzodiazepines interact primarily with brain GABAA
recep-tors in which the α subunits (isoforms 1, 2, 3, and 5) have a conserved
histidine residue in the N-terminal domain. Mice in which a point mutation has
been inserted converting histidine to arginine in the α1 subunit show
resistance to both the sedative and amnestic effects of benzodiazepines, but
anxi-olytic and muscle-relaxing effects are largely unchanged. These animals
are also unresponsive to the hypnotic actions of zolpidem and zaleplon, drugs
that bind selectively to GABAA receptors containing α1 subunits. In
contrast, mice with selec-tive histidine-arginine mutations in the α2 or α3
subunits of GABAA receptors show selective resistance to the
antianxiety effects of benzodiazepines. Based on studies of this type, it has
been suggested that α1 subunits in GABAA receptors mediate sedation,
amnesia, and ataxic effects of benzodiazepines, whereas α2 and α3 subunits are
involved in their anxiolytic and muscle-relaxing actions. Other mutation
studies have led to suggestions that an α5 subtype is involved in at least some
of the memory impairment caused by benzodiazepines. It should be emphasized
that these studies involving genetic manipulations of the GABAA
receptor utilize rodent models of the anxiolytic and amnestic actions of drugs
The Versatility of the Chloride Channel GABA Receptor Complex
The GABAA-chloride channel macromolecular
complex is one of the most versatile drug-responsive machines in the body. In
addition to the benzodiazepines, barbiturates, and the newer hypnotics (eg,
zolpidem), many other drugs with central nervous system effects can modify the
function of this important iono-tropic receptor. These include alcohol and
certain intravenous anesthetics (etomidate, propofol) in addition to
thiopental. For example, etomidate and propofol
appear to act selectively at GABAA receptors that contain β2
and β3 subunits, the latter suggested to be the most important with respect to
the hypnotic and muscle-relaxing actions of these anesthetic agents. The
anesthetic steroid alphaxalone is thought to interact with GABAA
receptors, and these recep-tors may also be targets for some of the actions of
volatile anesthetics (eg, halothane). Most of these agents facilitate or mimic
the action of GABA. However, it has not been shown that all these drugs act
exclusively by this mechanism. Other drugs used in the management of seizure
disorders indirectly influence the activity of the GABAA-chloride
channel macro-molecular complex by inhibiting GABA metabolism (eg, vigabatrin)
or reuptake of the transmitter (eg, tiagabine). Central nervous system
excitatory agents that act on the chloride channel include picrotoxin and
bicuculline. These convulsant drugs block the channel directly (picrotoxin) or
interfere with GABA binding (bicuculline).
D. Organ Level Effects
1.
Sedation—Benzodiazepines,
barbiturates, and most oldersedative-hypnotic drugs exert calming effects with
concomitant reduction of anxiety at relatively low doses. In most cases,
how-ever, the anxiolytic actions of sedative-hypnotics are accompa-nied by some
depressant effects on psychomotor and cognitive functions. In experimental
animal models, benzodiazepines and older sedative-hypnotic drugs are able to
disinhibit punishment-suppressed behavior. This disinhibition has been equated
with antianxiety effects of sedative-hypnotics, and it is not a charac-teristic
of all drugs that have sedative effects, eg, the tricyclic antidepressants and
antihistamines. However, the disinhibition of previously suppressed behavior
may be more related to behav-ioral disinhibitory effects of sedative-hypnotics,
including euphoria, impaired judgment, and loss of self-control, which can
occur at dosages in the range of those used for management of anxiety. The
benzodiazepines also exert dose-dependent antero-grade amnesic effects
(inability to remember events occurring during the drug’s duration of action).
2.
Hypnosis—By definition, all of
the sedative-hypnoticsinduce sleep if high enough doses are given. The effects
of sedative-hypnotics on the stages of sleep depend on several factors,
including the specific drug, the dose, and the frequency of its administration.
The general effects of benzodiazepines and older sedative-hypnotics on patterns
of normal sleep are as fol-lows: (1) the latency of sleep onset is decreased
(time to fall asleep); (2) the duration of stage 2 NREM (nonrapid eye
move-ment) sleep is increased; (3) the duration of REM sleep is decreased; and
(4) the duration of stage 4 NREM slow-wave sleep is decreased. The newer
hypnotics all decrease the latency to persistent sleep. Zolpidem decreases REM
sleep but has minimal effect on slow-wave sleep. Zaleplon decreases the latency
of sleep onset with little effect on total sleep time, NREM, or REM sleep.
Eszopiclone increases total sleep time, mainly via increases in stage 2 NREM
sleep, and at low doses has little effect on sleep patterns. At the highest
recommended dose, eszopiclone decreases REM sleep.
More
rapid onset of sleep and prolongation of stage 2 are presumably clinically
useful effects. However, the significance of sedative-hypnotic drug effects on
REM and slow-wave sleep is not clear. Deliberate interruption of REM sleep
causes anxiety and irritability followed by a rebound increase in REM sleep at
the end of the experiment. A similar pattern of “REM rebound” can be detected
following abrupt cessation of drug treatment with older sedative-hypnotics,
especially when drugs with short durations of action (eg, triazolam) are used
at high doses. With respect to zolpidem and the other newer hypnotics, there is
little evidence of REM rebound when these drugs are discontinued after use of
recommended doses. However, rebound insomnia occurs with both zolpidem and
zaleplon if used at higher doses. Despite possible reductions in slow-wave
sleep, there are no reports of disturbances in the secretion of pituitary or
adrenal hormones when either barbiturates or benzodiazepines are used as
hypnotics. The use of sedative-hypnotics for more than 1–2 weeks leads to some
tolerance to their effects on sleep patterns.
3.
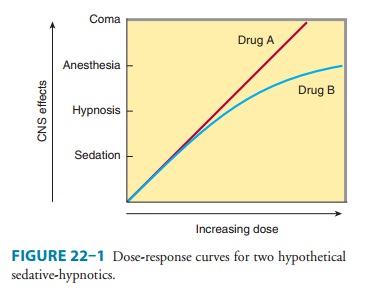
Anesthesia—As shown in Figure
22–1, high doses of certainsedative-hypnotics depress the central nervous
system to the point known as stage III of general anesthesia . However, the
suitability of a particular agent as an adjunct in anesthesia depends mainly on
the physicochemical properties that determine its rapidity of onset and
duration of effect. Among the barbiturates, thiopental and methohexital are
very lipid-soluble, penetrating brain tissue rapidly following intravenous
administra-tion, a characteristic favoring their use for the induction of
anes-thesia. Rapid tissue redistribution (not rapid elimination) accounts for
the short duration of action of these drugs, a feature useful in recovery from
anesthesia.
Benzodiazepines—including
diazepam, lorazepam, and midazolam—are used intravenously in anesthesia , often
in combination with other agents. Not surprisingly, benzodiazepines given in
large doses as adjuncts to general anes-thetics may contribute to a persistent
postanesthetic respiratory depression. This is probably related to their
relatively long half-lives and the formation of active metabolites. However, if
neces-sary, such depressant actions of the benzodiazepines are usually
reversible with flumazenil.
4.
Anticonvulsant effects—Many
sedative-hypnotics arecapable of inhibiting the development and spread of
epileptiform electrical activity in the central nervous system. Some
selectivity exists in that some members of the group can exert anticonvulsant
effects without marked central nervous system depression (although psychomotor
function may be impaired). Several benzodiazepines—including clonazepam,
nitrazepam, lorazepam, and diazepam—are sufficiently selective to be clinically
useful in the management of seizures . Of the barbiturates, phenobarbital and
metharbital (converted to phenobarbital in the body) are effective in the
treatment of generalized tonic-clonic seizures, though not the drugs of first
choice. Zolpidem, zaleplon, and eszopiclone lack anticonvulsant activity,
presumably becauseof their more selective binding than that of benzodiazepines
to GABAA receptor isoforms.
5. Muscle relaxation— Some sedative-hypnotics,
particularlymembers of the carbamate (eg, meprobamate) and benzodiazepine
groups, exert inhibitory effects on polysynaptic reflexes and inter-nuncial
transmission and at high doses may also depress transmis-sion at the skeletal
neuromuscular junction. Somewhat selective actions of this type that lead to
muscle relaxation can be readily demonstrated in animals and have led to claims
of usefulness for relaxing contracted voluntary muscle in muscle spasm (see
Clinical Pharmacology). Muscle relaxation is not a characteristic action of
zolpidem, zaleplon, and eszopiclone.
6.
Effects on respiration and
cardiovascular function— Athypnotic doses in healthy patients, the effects of
sedative-hypnotics on respiration are comparable to changes during natural sleep.
However, even at therapeutic doses, sedative-hypnotics can pro-duce significant
respiratory depression in patients with pulmonary disease. Effects on
respiration are dose-related, and depression of the medullary respiratory
center is the usual cause of death due to overdose of sedative-hypnotics.
At
doses up to those causing hypnosis, no significant effects on the
cardiovascular system are observed in healthy patients. However, in hypovolemic
states, heart failure, and other diseases that impair cardiovascular function,
normal doses of sedative-hypnotics may cause cardiovascular depression,
probably as a result of actions on the medullary vasomotor centers. At toxic
doses, myocardial contractility and vascular tone may both be depressed by
central and peripheral effects, leading to circulatory collapse. Respiratory
and cardiovascular effects are more marked when sedative-hypnotics are given
intravenously.
Related Topics