Chapter: Pharmaceutical Biotechnology: Fundamentals and Applications : Interferons and Interleukins
Pegylated Interferons and Interleukins: The Next Generation
PEGYLATED INTERFERONS AND INTERLEUKINS: THE NEXT GENERATION
Since 1977 it has been known that PEG conjugated proteins are frequently more effective than their native parent molecule. Our understanding of PEG chemistry and how it affects the behavior of a biopharmaceutical has increased with the number of PEGylated proteins developed as therapeutic agents (Table 5 gives some examples). PEG is inert, nontoxic, nonimmunogenic and in its most common form either linear or branched terminated with hydroxyl groups that can be activated to couple to the desired target protein. It has been approved for human administration by mouth, injection, and topical application. Its general structure is:
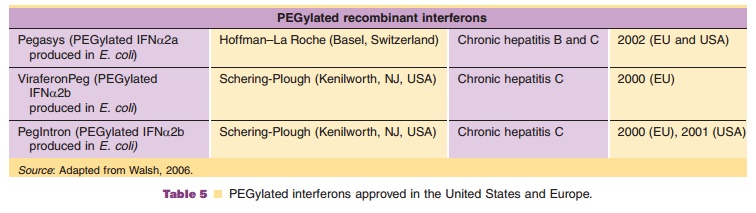
Bifunctional linear PEG (diol):
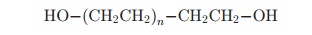
For polypeptide modification one hydroxyl group is usually inactivated by conversion to mono-methoxy or mPEG and becomes monofunctional, i.e., only one hydroxyl group is activated during the PEGylation process, thus avoiding the formation of interprotein (oligomerization) or intraprotein bridges:
Monofunctional linear mPEG:
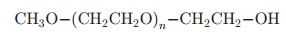
To couple PEG to a molecule such as polypep-tides, polysaccharides, polynucleotides or small or-ganic molecules it is necessary to chemically activate it. This is done by preparing a PEG derivative with a functional group chosen according to the desired profile for the final product. In addition to the linear PEGs, branched structures have proven useful for peptide and protein modifications:
Branched PEG:
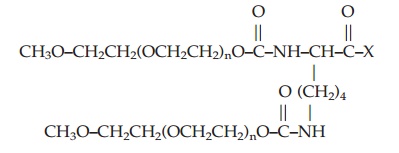
Branched PEG or PEG2 have a number of advantages over linear structures:
· Attached to proteins they “act” much larger than a linear mPEG of the same MW.
· Two PEG chains are added per attachment site, reducing the chance of protein inactivation.
· More effective in protecting proteins from proteo-lysis, reducing antigenicity and immunogenicity.
Depending on the desired use for the PEG-modified molecule different PEGylation strategies can be chosen, for example:
· Multiple shorter chain PEGylation if the biological activity should be preserved.
· A weak PEG-protein bond if a slow release effect is desired.
· A branched chain with high MW and a strong bond if prolonged circulation and receptor satura-tion is the goal.
Table 5 lists some of the PEGylated protein pharmaceuticals on the market or in various phases of development with appropriate references. For a more in-depth review of PEG chemistries and characteristics the interested reader is referred to Roberts et al. (2002) and Bailon et al. (2001).
The development of rhIFNα from the native, unmodified molecule to the PEGylated form with the desired pharmacological profile is an example of how the understanding of PEG chemistry progressed with ex-perience (Zeuzem et al., 2003). Increasing the length of the PEG chain resulted in progressively longer circulating half-life due to protracted resorption and lower clearance, ultimately resulting in a near constant serum concentration over an entire week summarized in Figure 3.
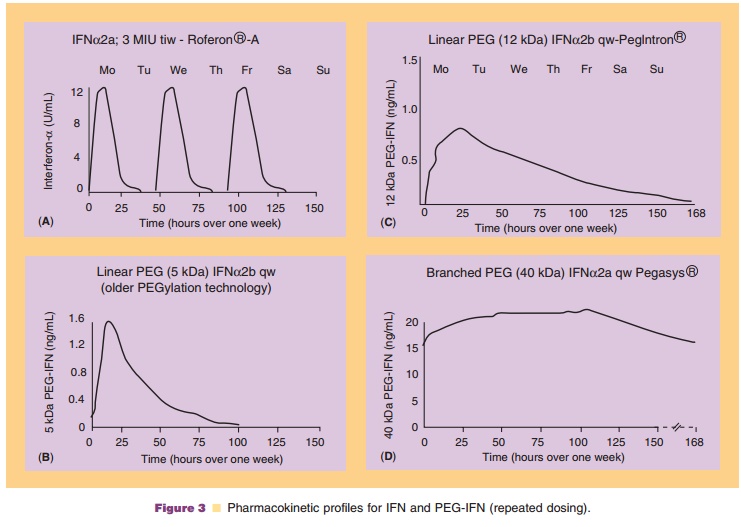
The first PEGylated IFN, IFNα2a, used a linear, 5 kDa mPEG with a weak urethane PEG-IFNα2a link. Clinical trials conducted with this compound were unsuccessful because the blood circulation half-life for the conjugate (Fig. 3B) was only slightly improved relative to that of the native protein (Fig. 3A) (Wills, 1990). Development of the product was therefore halted at Phase II clinical trials (Zeuzem et al., 2003). The second compound was developed by Schering Plough, Kenilworth, New Jersey in collaboration with Enzon Pharmaceutical Inc, Bridgewater, New Jersey. It made use of a longer (12 kDa), linear PEG with an urethane linkage to IFNα2b. The chosen strategy was to combine the advantages of high specific activity with slower serum clearance resulting in PegIntron (Wang et al., 2002) with markedly improved pharmacological properties allowing once a week administration (Fig. 3C) (Glue et al., 2000). PegIntron, also marketed as Viraferon in some countries, is approved world-wide for the treatment of chronic hepatitis C.
The development of the third PEGylated IFN, IFNα2a, took a different approach. The strategic goal was to achieve lasting and constant serum concentra-tions over an entire week. In a collaboration of Roche with Shearwater Polymers in Huntsville, Alabama (now Nektar; San Carlos, California), IFNα2a was linked by a stable amide bond to four different PEG chains of various sizes, structures, and site-attachment numbers. The resulting products were tested for antiviral activity and a variety of pharmacokinetic parameters including half-life, absorption rate, and mean residence time:
· 20-kDa linear mono-PEGIFNα2a,
· 40-kDa linear di-PEGIFNα2a,
· 20-kDa branched mono-PEGIFNα2a
· 40-kDa branched mono-PEGIFNα2a
The 40-kDa, branched PEGylated molecule (later named Pegasys) exhibited sustained absorption, decreased systemic clearance, and an approximate 10-fold increase in serum half-life over regular IFN. The biological activity was similarly prolonged resulting in an optimal pharmacological profile, Figure 3D (Algranati et al., 1999). It was therefore chosen for further clinical development (Reddy et al., 2002) leading to its approval worldwide for the treatment of chronic hepatitis B and C. Pegasys is being tested for the treatment of renal cell carcinoma (Motzer et al., 2002), malignant melanoma, chronic myeloid leukemia (Talpaz et al., 2005) and NHL.
The rapidly growing understanding of the potential of advanced PEGylation chemistry to improve the stability and pharmacological properties of biopharmaceuticals has fostered the development of an increasing number of PEG-biopharmaceuticals. Several of those have proven to offer significant advantages over their native counterparts and found their place in our therapeutic armamentarium. PEG is also used for a variety of other (nonbio) pharmaceutical applications. Table 6 lists several examples of different marketed products and others still in development.
Related Topics