Chapter: Pharmaceutical Drug Analysis: Iodimetric and Iodometric Titrations
Iodimetric Assays
IODIMETRIC ASSAYS
In such estimations, the pharmaceutical substances can be
measured either directly or back titration of excess iodine with sodium
thiosulphate solution.
1. Direct Titration with Iodine
(a) Preparation of 0.1 Iodine Solution
Theory : Iodine in aqueous solution
acts as an oxidizing agent which forms the basis of assay methods involving direct titration with
iodine. Thus, we have :
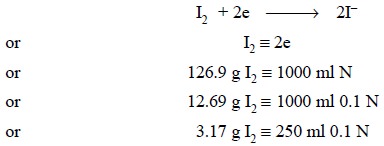
Materials Required : Iodine : 3.2 g ; potassium
iodide : 7.5 g.
Procedure : Weigh accurately 3.2 g of
crushed iodine crystals on a watch glass and transfer to a beaker containing potassium iodide (7.5 g)
and water (10 ml). Dissolve the contents of the beaker with the help of a glass
rod and frequent swirling. Transfer the contents of the beaker quantitatively
to a 250 ml volumetric flask and make up the volume with DW.
Explanation : Iodine is sparingly soluble in
water but undergoes rapid dissolution in the presence of potassium iodide due to the formation of the corresponding
triiodide ion :
I2 + I–
→ I3–
Thus, potassium iodide plays dual role, viz., in iodimetry—to solubilize iodine
in aqueous KI solution, and in iodometry—as reducing agent, the excess KI helps
in retaining liberated I2 in solution through interaction with KI.
(b) Standardization of 0.1 Iodine Solution with
the aid of Arsenic Trioxide (As2O3)
Theory : This particular
standardization is solely governed by the following equations, namely :
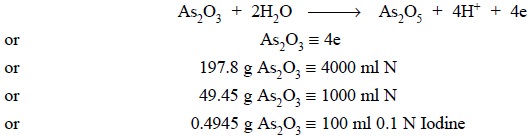
Hydroiodic acid (HI) possesses strong reducing
characteristics which renders the oxidation with iodine into a reversible
reaction as follows :

In order to shift the equilibrium to the right-hand-side
(i.e., towards As2O5)
in the above reaction, sodium bicarbonate (NaHCO3) is employed to
remove the HI generated. It is important to record here that neither sodium
hydroxide nor sodium carbonate can be used as both of them produce sodium
iodide (NaI) and sodium iodate (NaIO3) as designated below :
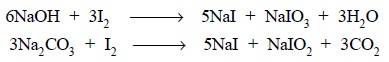
Materials Required : Arsenic trioxide : 0.5 g ;
sodium hydroxide solution (20% w/v in water) : 2 ml ; dilute hydrochloric acid
(2N) ; sodium bicarbonate : 4 g ; 0.1 N iodine solution.
Procedure : Weigh accurately 0.5 g arsenic
trioxide into a beaker, add to it 2 ml of sodium hydroxide solution, and heat to dissolve. Cool and transfer the contents
quantitatively to a 100 ml volumetric flask and make up the volume upto the
mark with DW. Pipette 20 ml into an iodine-flask, acidify with dilute HCl
carefully and confirm it by adding a little NaHCO3 to remove the
free excess acid, followed by a further 2 g to get rid of HI formed in the
reaction mixture. Now, titrate with 0.1 N iodine solution till the end-point is
achieved by the appearance of the first permanent pale straw colour.
(c) Standardization of 0.1 Iodine Solution by
the aid of Sodium Thiosulphate
Theory : Iodine solution may also be
standardized by using sodium thiosulphate (AR-Grade) whereby the latter gets oxidized to sodium
tetrathionate as expressed below :
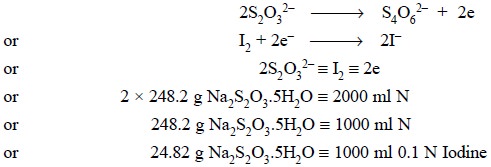
Materials Required : Sodium thiosulphate (AR) :
6.025 g ; 0.1 N I2 solution.
Procedure : Weigh accurately 6.025 g of
sodium thiosulphate (AR) to a 250 ml volumetric flask. Dissolve it in DW, shake well and make up the volume to the mark
with DW. Pipette 25 ml of 0.1 iodine solution into an iodine flask and titrate
with the standard sodium thiosulphate solution (as primary standard) until the
solution becomes almost colourless.
Note : Stock solutions of
sodium thiosulphate may be preserved by the addition of a few drops of sodium
hydroxide solution (20% w/v) which serves as stabilizer as well as prevents
decomposition.
(d) Preparation of Starch Solution
Material Required : Starch (arrowroot) : 1.0 g.
Procedure : Weigh 1.0 g starch in a glass
in a glass pestle-mortar and triturate thoroughly with 10 ml of cold DW. Boil separately 200 ml of
DW in a beaker and add the starch paste to it with vigorous stirring. The
resulting mixture is boiled gently for a further period of 30 minutes till a
transluscent and thin liquid having an uniform consistency is obtained.
Note : (1) The prepared
solution of starch undergoes rapid deterioration, hence it is always desired to
use freshly prepared solution every day,
(2) It is now more or
less believed that the iodine is held as an ‘absorption complex’ within the
helical chain of the macromolecule β-amylose i.e., a component of most starches. However, another component, α-amylose, is undesirable because it produces a
red-colouration with io-dine which is not readily reversible, and
(3) ‘Soluble Starch’
comprises principally of β-amylose, with the α-fraction having been removed. Always, it is a practice
to prepare indicator-solutions from this product exclusively.
1.1. Analgin
Materials Required : Analgin : 0.4 g ; alcohol
(95%) : 40 ml ; 0.01 N hydrochloric acid : 10 ml ; 0.1 N iodine solution.
Theory : The estimation of analgin
depends upon the oxidation of the enolic group with iodine. The reaction is not reversible :
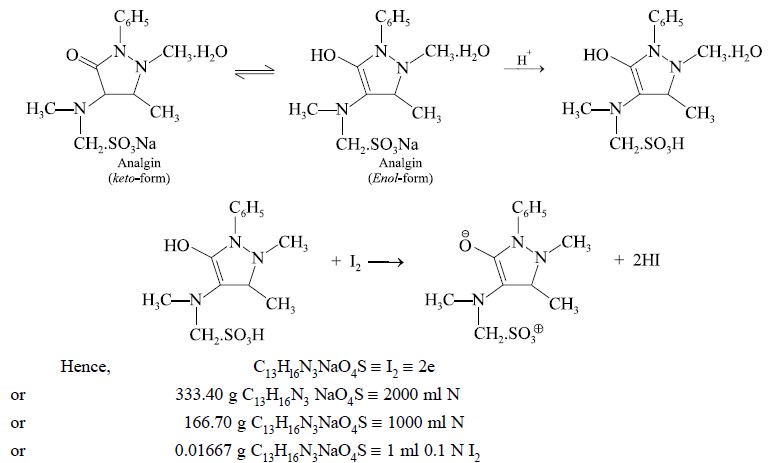
Procedure : Weigh accurately about 0.4 g
and dissolve in a mixture of 40 ml of alcohol and 10 ml of 0.01 N hydrochloric acid. Titrate the resulting mixture with 0.1 N
iodine solution till a yellow colour that remains stable for 30 seconds is achieved.
Each ml of 0.1 N iodine is equivalent to 0.016670 g of C13H16N3NaO4S.
1.2. Acetarsol
Materials Required : Acetarsol : 0.25 g ; sulphuric
acid (conc.) : 7.5 ml ; nitric acid (fuming) : 2.5 ml ; ammonium sulphate : 5 g ; potassium iodide : 1.0 g ; sodium
sulphite (0.1 N) : 1.0 ml ; phenolphthalein solution : 2 drops ; NaOH solution
(0.1 N) ; dilute sulphuric acid (6 N) ; sodium bicarbonate : 8.0 g ; iodine
solution (0.1 N).
Theory : Acetarsol is an organic
arsenal, hence arsenic may be estimated by carrying out the oxidation As3+ to As5+
state with the help of 0.1 N iodine solution.
The organic entity present in acetarsol is destroyed
primarily by boiling it with aqua-regia (a mixture of conc. H2SO4
and fuming nitric acid). The resulting mixture is heated in the presence of
ammonium sulphate to get rid of nitric
acid finally in the form of nitrous oxide (N2O) as follows :
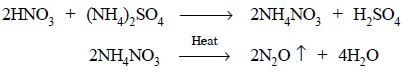
Previously added H2SO4 maintains an
acidic medium which on adding KI liberates HI that reduces the As5+
to As3+ state. Reduction is completed by boiling the solution which
also expels the liberated I
as shown below :
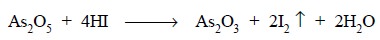
The resulting mixture is cooled to room temperature and
the residual iodine is removed by titration with 0.1 N sodium sulphite
solution. Now, the solution is treated with sodium hydroxide solution to make
it alkaline and then acidified carefully with dilute H2SO4
to remove the free NaOH. Finally, the resulting solution is made alkaline with
NaHCO3 so that the equilibrium is shifted to the right (i.e., AS3+ gets converted to
As5+) quantitatively on carrying out the titration with 0.1 N iodine
solution. Thus, we have :
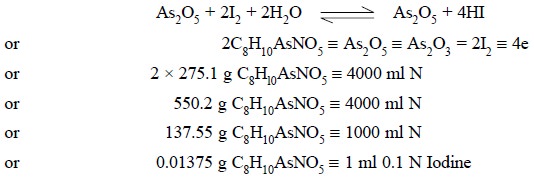
Procedure : Weigh accurately about 0.25 g
of acetarsol into a 500 ml iodine flask and add to it sulphu-ric acid (conc.)
7.5 ml, followed by nitric acid (fuming) 1.5 ml. Boil the contents of the flask
gently for 45 minutes preferably in a fume-cupboard. Cool the solution, add 0.5
ml of fuming HNO3 and boil till brown vapours (N2O) stop
coming. Again cool the contents and add carefully 5 g of ammonium sulphate in
small lots at intervals and heat till there is no evolution of N2O
thereby giving rise to a colourless liquid. Bring the solution to room
temperature, dilute with 100 ml DW, add 1 g KI and heat gently till the volume
becomes 50 ml. Cool and add a few drops of 0.1 N sodium sulphite to effect
decolourisation. Add 60 ml DW to dilute the resulting contents and make it just
alkaline with NaOH solution by adding phenolphthalein indicator. Finally,
acidify with dilute H2SO4, neutralize with NaHCO3
and add 4 g of NaHCO3 in excess. Swirl the contents of the flask and
titrate with 0.1 N iodine solution. Each ml of 0.1 N iodine solution is
equivalent to 0.01375 g of C8H10AsNO5.
1.3. Cognate Assays
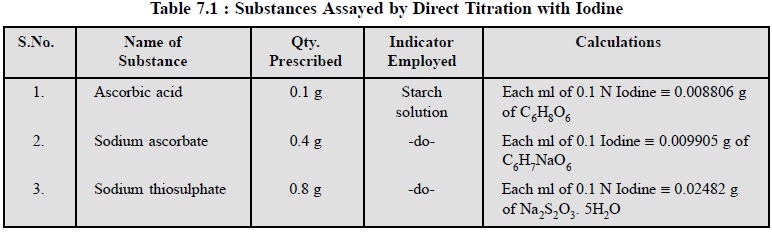
The following pharmaceutical substances can be assayed by
direct titration with iodine as stated in Table 7.1.
2. Residual Titration Method (Excess of Iodine Titrated with Sodium Thiosulphate)
In this titration method an excess of iodine solution is
added to the solution of the substance and thus, the latter gets oxidized
quantitatively. The excess of iodine is subsequently back titrated with sodium
thiosulphate using freshly prepared starch solution as indicator with an end-point
from violet to colourless.
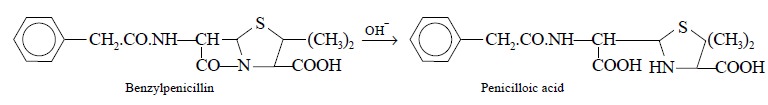
2.1. Benzylpenicillin
Theory : Benzylpenicillin can be
assayed efficiently by adopting the following three steps sequentially, namely
:
Step 1 : Benzylpenicillin is first
converted to the corresponding penicilloic acid (a dicarboxylic acid) by
carrying out the hydrolysis with sodium hydroxide solution, as follows :
Step 2 : Penicilloic acid on treatment
with acid yields D-penicillamine and benzylpenilic acid, as shown under :
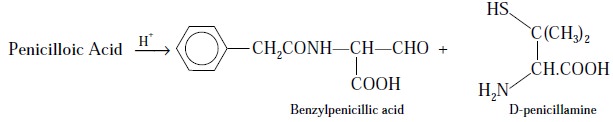
Step 3 : D-Penicillamine thus obtained
is oxidised quantitatively by iodine to give rise to a disulphide, as expressed in the following equation
; whereas, the excess iodine is back titrated with 0.02 N sodium thiosulphate
solution :
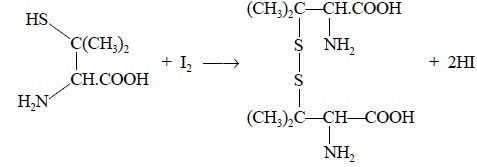
From the above reaction, we have :
C16Hl7N2NaO4S
≡ I ≡ e
In usual practice, however, benzylpenicillin sodium is
standardised against a chemical reference sub-stance of pre-determined potency.
Materials Required : Benzylpenicillin : 0.1 g ; (N)
sodium hydroxide solution : 5 ml ; buffer solution (5.44% w/v of CH3COONa
and 2.40% w/v of
glacial acetic acid) : 20 ml ; (N) hydrochloric acid : 5 ml ; 0.02 N iodine
solution : 25 ml ; 0.02 N sodium thiosulphate solution ; starch solution.
Procedure : Weigh accurately about 0.1 g
of benzylpenicillin in DW and dilute to 100 ml in a volumet-ric flask. Transfer
10.0 ml to an iodine flask, add 5 ml of N sodium hydroxide and allow to stand
for 20 minutes. Now, add 20 ml of freshly prepared buffer solution, 5 ml of N
HCl and 25.0 ml of 0.02 N iodine solution. Close the flask with a wet
glass-stopper and allow to stand for 20 minutes in a dark place (i.e., protected from light). Titrate the
excess of iodine with 0.02 N sodium thiosulphate, employing freshly pre-pared
starch solution as an indicator added towards the end-point.
To another 10.0 ml of the initial solution add 20 ml of
the buffer solution, allow to stand for 20 minutes in the dark and titrate with
0.02 N sodium thiosulphate, using starch solution, added towards the end of the
titration as indicator.
The difference between the two titrations represents the
volume of 0.02 N iodine equivalent to the total penicillins present in the
given sample of benzylpenicillin. An assay may be carried out simultaneously by
benzylpenicillin sodium (reference sample) so as to determine the exact
equivalent of each ml of 0.02 N iodine.
Calculations : Calculate the potency in Units
of penicillin from the declared number of Units of pencillin in benzylpenicillin sodium (reference sample).
2.2. Sodium Metabisulphite
Theory : Sodium metabisulphite in
acidic medium (HCl) yields SO2
which reacts with water to pro-duce sulphurous acid. The generated
sulphurous acid is quantitatively oxidized by iodine to sulphuric acid, and the
excess iodine is subsequently back titrated with sodium thiosulphate. The
various reactions can be expressed as shown below :
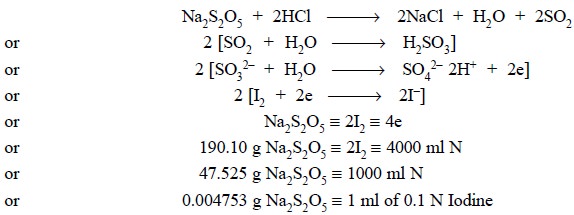
Materials Required
: Sodium metabisulphite
: 0.2 g ; 0.1 N Iodine solution ; hydrochloric acid ( ~− 11.5 N) : 1 ml ; 0.1 N sodium thiosulphate ;
starch solution.
Procedure : Weigh accurately about 0.2 g
of sodium metabisulphite and dissolve in 50.0 ml of 0.1 N iodine solution and add 1 ml hydrochloric acid. Titrate the excess
of iodine with 0.1 N sodium thiosulphate employing freshly prepared starch
solution, added towards the end of the titration, as indicator. Each ml of 0.1
N iodine is equivalent to 0.0047453 g of Na2S2O5.
2.3. Cognate Assays
A few other pharmaceutical substances may also be assayed
by adopting the residual titration method as shown in Table 7.2.

Related Topics