Chapter: Pharmaceutical Drug Analysis: Infrared Spectrophotometry
Infrared Spectrophotometry: Theory
THEORY
The underlying principle of infrared spectroscopy is
based upon the molecular vibrations which is further composed of the stretching
and the bending vibrations of a molecule.
Therefore, it would be necessary to have a clear concept
of various modes of vibrations often encoun-tered in different molecules having
a variety of functional moieties, laws governing them and the mathemati-cal
derivations related to them.
1. MOLECULAR VIBRATIONS
A molecule may not be looked upon as a rigid assemblage
of atoms. Rather it may be regarded as a sort of flexible system comprising of
balls of varying masses representing the atoms of a molecule and springs of
varying strengths representing the chemical bonds of a molecule.
The vibrations for molecules are of two types, namely :
(a) Stretching,
and
(b) Bending (or deformation).
1.1. Stretching
Vibration causes stretching whereby the distance between
the two atoms increases or decreases, but the atoms remain in the same bond
axis.
1.2. Bending (or Deformation)
Vibration causes bending whereby the position of the atom
changes relative to the original bond axis.
Therefore, the various stretching and bending vibrations
of a bond usually take place at particular quantized frequencies. Thus, in a
situation where upon the infrared light having the same frequency is inci-dent
on the molecule, energy is absorbed, and the net effect could be observed by an
increase in the ampli-tude of that vibration. In another situation, whereby the
molecule reverts from the excited state to the ground state, the absorbed
energy is released in the form of heat.
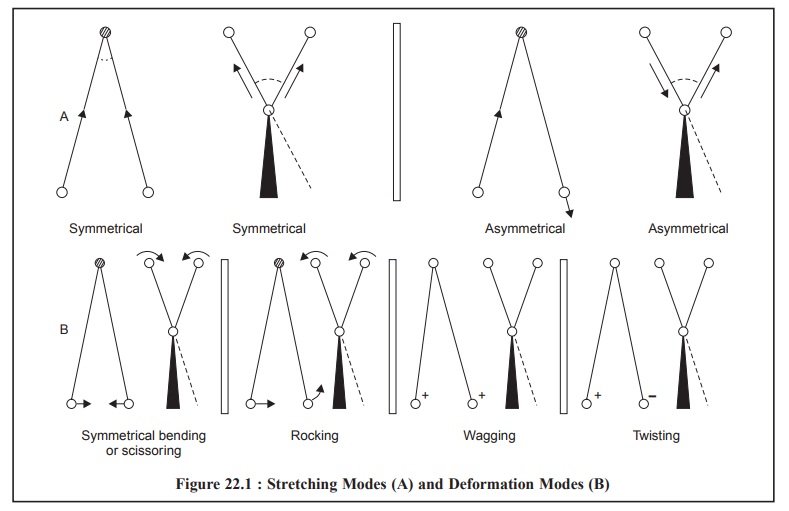
The various stretching and bending vibrations that can
exist within a molecule may be represented schematically as shown below in
Figure 22.1 :
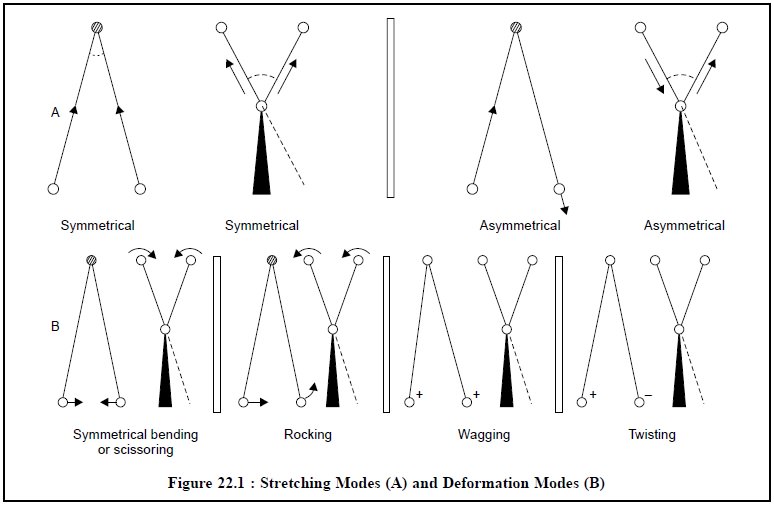
There are two
types of Bending (or deformation) Modes, namely :
(i) Below the
plane of paper and perpendicular to it designated by (+) sign, and
(ii) Above the
plane of paper and perpendicular to it represented by (–) sign.
1.3. Stretching Vibrations
In this particular instance, the atoms move invariably
along the bond that joins them e.g.,
C—H ; C = O ; O—H ; N—H.
The stretching vibrations may be further sub-divided into
two categories, namely :
(a) Symmetrical Stretching : In this case, the two
hydrogen atoms either move towards or away from the central carbon atom in
unison, thereby either altering the interatomic distance or causing no change
in valence angle (Figure 22.1).
(b) Asymmetrical Stretching : In this
instance, one hydrogen atom approaches the carbon atom while the other moves
away from the carbon atom (Figure 22.1).
1.4. Bending (or Deformation) Vibrations
In the event when a three-atom system forms part of a
larger molecule, it is quite possible to have bending (or deformation)
vibrations which essentially involve oscillation of the atoms, or group as a
whole and is perpendicular to its chemical bond (Figure 22.1).
Such bending vibrations can take place either in-plane or
out-of-plane.
1.4.1. In-Plane Bending Vibrations
These are two
types :
(a) Scissoring or Symmetrical Bending : In
this case, the two atoms connected to a central atom either move toward or away
from each other with certain deformation of the valence angle.
(b) Rocking : In this case, the structural
unit swings back and forth in the plane of the molecule.
1.4.2. Out-of Plane Bending
Vibrations
These are also of two
kinds, namely :
(a) Wagging : In this case the structural
unit swings back and forth out of the plane of the molecule.
(b) Twisting : In this case the structural
unit rotates about the bond that joins it to the rest of the molecule.
1.4.3. Explanations of Bending and
Stretching Vibrations
The bending (or deformation) vibrations generally require
less energy and take place at longer-wave-length than the corresponding
stretching vibrations.
In contrast, the stretching vibrations are observed to
occur with respect to their corresponding bond-strengths.
Examples : The typical examples of
triple-bond, double-bond and single-bond are given below :
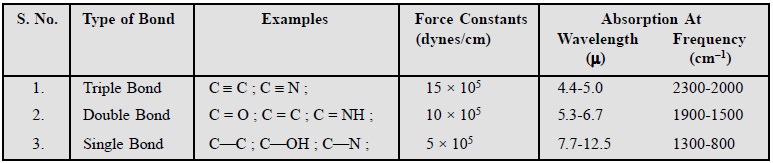
Whenever a very small proton like : C—H ; O—H ; or N—H is
involved in a single bond, the stretch-ing vibrations normally take place at
much higher frequency i.e., 3700-2630
cm–1 (or 2.7-3.8 µ). It is, how-ever,
interesting to note that O—H bond absorbs at 2.8 µ
(or 3570 cm–1), whereas O—D bond absorbs at 3.8μ (or 2630 cm–1). In this specific case, the
strengths of the two bonds are more or less the same, but the mass of one atom
is almost doubled.
1.4.4. Calculation of Vibrational
Frequencies
The vibrational frequency may be calculated with fairly
remarkable accuracy by the help of Hooke’s Law and is expressed as :
 ...............................(a)
...............................(a)
Where , ν =
Frequency,
k = Force constant of the bond,
and
m1 and m2 = Masses of two atoms.
The quantity m1m2/(m1 + m2)
is often expressed as µ, the reduced mass of the
system.
Example : Calculate the approximate
frequency of the C—H stretching vibration from the following dara:
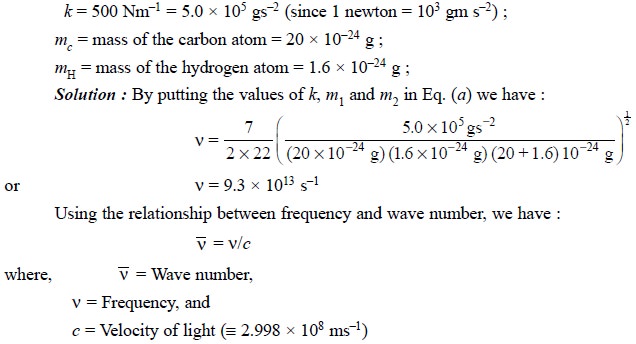
Therefore, we have :
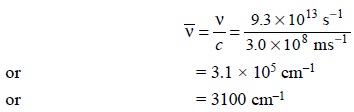
However, it is pertinent to restate the underlying
principles embodied in these calculations, that is “the vibrational frequency of a bond is expected to increase when the
bond strength increases, and also when the reduced mass of the system
decreases”.
1.5. Salient Features of IR-Spectroscopy
There are, in fact, three
vital points that may be noted with regard to IR-spectroscopy, namely :
(a) Most
intense peaks in the IR-spectrum are solely due to absorption peaks caused by
the stretching vibrations,
(b) Molar
extinction coefficient in IR-spectroscopy varies from 0-2000 cm–1,
and
(c) Molar
extinction coefficient is directly proportional to the square of the change in
the dipole moment* of the molecule that the particular vibration affords.
2. FACTORS INFLUENCING VIBRATIONAL FREQUENCIES
There are a number of factors that influence the precise
frequency of a molecular vibration, namely :
(a) Vibrational
coupling,
(b) Hydrogen bonding,
(c) Electronic
Effects, and
(d) Field
Effects.
All the above factors shall be discussed briefly with
appropriate examples and explanations, wherever necessary, below :
2.1. Vibrational Coupling
The following four vibrations may be observed in the
high-resolution spectra of compounds contain-ing both —CH2 and —CH3
groups.
C—H : One stretching
frequency,

Examples : The three typical examples
are described below, namely :
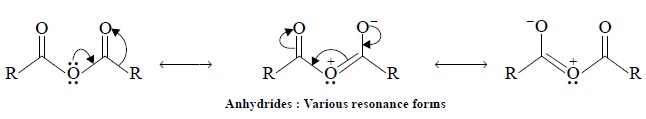
(a) Carboxylic Acid
Anhydrides : For instance :

Following are the salient features :
·
It affords two C = O stretching absorptions viz., νanti and νsymm between frequency 1800-1900
cm–1 having a separation of about 65 cm–1 ;
·
The coupling solely occurs between the two carbonyl
groups, that are indirectly linked through —O— ;
·
Resonance : The interaction is probably
encouraged due to the single as well as double-bond character prevailing in the carbonyl-oxygen bonds brought about by
resonance as shown below :
(b) Amides  : The functional moiety
‘amide’ shows two distinct bands around 1600- 1700 cm–1 caused due
to C = O str. and N—H def. These bands are also known as Amide-I and Amide-II
bands.
: The functional moiety
‘amide’ shows two distinct bands around 1600- 1700 cm–1 caused due
to C = O str. and N—H def. These bands are also known as Amide-I and Amide-II
bands.
Amide-I : is formed due to C = O str. and amounts to a level as high as
80% ;
Amide-II : is a strongly coupled
interaction between N—H def. and C—N str. and accounts for the remaining 20%.
(c) Aldehydes  : The functional group
‘aldehyde’ offers C—H str. absorption band which appears as a doublet because
of interaction between the two components, namely : C—H str. fundamental and
C—H def. overtone.
: The functional group
‘aldehyde’ offers C—H str. absorption band which appears as a doublet because
of interaction between the two components, namely : C—H str. fundamental and
C—H def. overtone.
2.2. Hydrogen Bonding
In general, the hydrogen bonding present in O—H and N—H
compounds give rise to a number of effects in the IR-spectra. However, the
carbonyl groups or aromatic rings present in the same molecule as the O—H or
N—H group may cause similar shifts by intramolecular action.
A few typical examples involving hydrogen bonding in a
wide-range of organic compounds are discussed below :
(a) Alcohols and Phenols : Interestingly,
alcohols and phenols, in condensed phases (KBr-Disc/ liquid film), are strongly
hydrogen-bonded thereby forming dimers, trimers and tetramers that ultimately
leads to a wide envelope of absorptions thus causing broadening of the
absorption band.
In diluted solution and in an inert solvent (e.g., 1-butanol 1% in CCl4)
the proportion of free molecules enhances that gives rise to the 3650 cm–1
band.
Another school of thought suggests that the hydrogen-bond
may be regarded as a resonance hy-brid I and II (approximating overall to
III)-thereby the original O—H bond undergoes a lengthen-ing as depicted below :
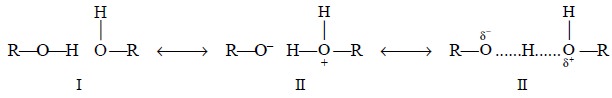
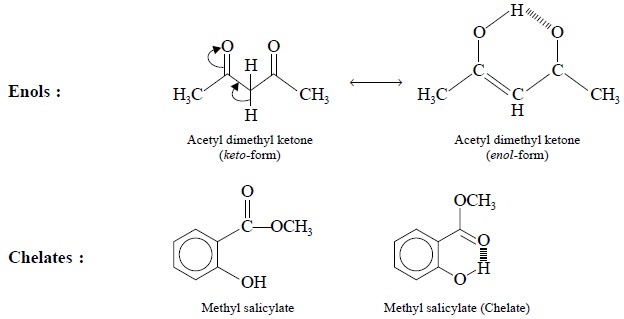
(b) Enols and Chelates : The
hydrogen-bonding existing in enols and chelates is found to be specifi-cally
strong and the observed O—H str. frequencies may be very low (2800 cm–1).
In this particu-lar instance, the dilution by an inert solvent (e.g., 1-butanol 1% in CCl4)
cannot even break these bonds, therefore, free O—H str. may not be observed at
low concentrations.
The above observations may be explained by citing the
example of acetyl dimethyl ketone (for enols) and methyl salicylate (for
chelates) as shown below :
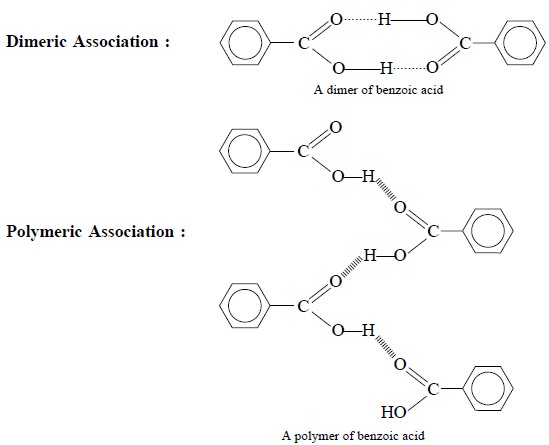
(c) Carboxylic Acids : Let us consider the
example of benzoic acid IR-spectrum in KBr-disc wherein we may observe a
relatively broad band caused due to hydrogen-bonded O—H str. between 2500-3500
cm–1. These broad bands are formed due to the dimeric and polymeric
associations of benzoic acid as shown below :
(d) π-Cloud Interactions : Since alkene and
aromatic π-bonds behave as Lewis bases, they may form hydrogen
bonds to acidic hydrogens e.g.,
Example : The frequency of O—H str. in
phenols is lowered by 40-100 cm–1 when the IR-spec-trum is
recorded in a benzene solution as compared to a carbon tetrachloride solution.
(e) Amines : Amines usually show two distinct bands due to N—H
stretching in different environ-ments. In condensed-phase spectra, amines show bonded N—H str. around 3300 cm–1
and in dilute solution a new band near 3600 cm–1 corresponding to
free N—H str. It may be attributed due to the fact that the electronegativity
of nitrogen is less than that of oxygen and hence the hydrogen bonds in amines
are weaker than in alcohols.
2.3. Electronic Effects
On the basis of theoretical principles one may explain
the frequency shifts that normally take place in molecular vibrations when the
substituents are altered :
A few such classical examples are enumerated with
appropriate explanations.
(a) Conjugation Effect : It is observed to lower the
frequency of both C = C str. and C = O str., irrespective of the fact it is
brought about by either αβ-unsaturation or by an
aromatic ring as shown below :
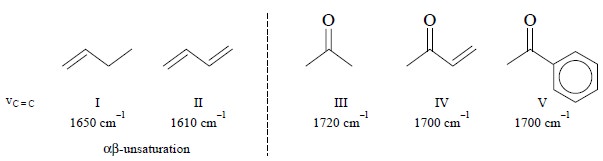
Explanation : In II, lowering of band order
in the C = C bond is observed due to conjugation effect whereby the stretching vibration frequency is decreased by
40-50 cm–1 (compare I and II above). In a similar manner, in V
delocalization of π-electrons between C = O and the benzene ring enhances
the double-bond character of the bond joining the C = O to the ring. It
ultimately leads to a lower band order in the C = O bond thereby decreasing the
stretching vibration fre-quency by 20-30 cm–1 (compare III and V
above).
(b) Mesomeric (or Resonance) Effect :
Whenever a molecule can be represented by two or more structures that differ
only in the arrangement of electrons—that is, by structures that have the same
arrangement of atomic nuclei-there is resonance.
It may be further expatiated with the help of the
following typical examples :
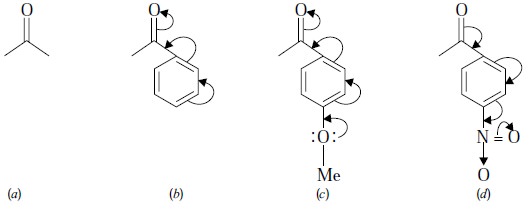
—OMe : Electron Releasing Moiety
—NO2 : Electron Withdrawing Moiety
In (b) above,
the presence of a phenyl ring increases the mesomeric
shift thereby lowering C = O str. frequency.
+M Group : viz., p-OMe (an electron releasing
function)—its presence as depicted in (c)
above will further lower frequencies
due to enhanced mesomeric effect.
– M Group : viz., p-NO2 (an electron withdrawing function)—its presence as shown in (d) above will further increase frequencies due to decreased mesomeric effect.
(c) Inductive Effects : The inductive effects solely
depends upon the ‘intrinsic’ tendency of a substituent to either release or
withdraw electrons-by definition, its electronegativity acting either through
the molecular chain or through space. This effect usually weakens steadily with
increas-ing distance from the substituent.
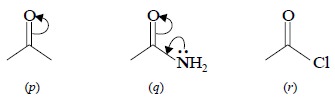
The ‘inductive effects’ shall now be discussed
specifically with regard to the various functional moieties such as : amides,
acyl chlorides, alkyl esters and aryl esters :
(i) Amides : (q), The + M effect causes weakening of the C = O bond, leading to
the corresponding ketone (p). In this
particular instance, the – I effect of nitrogen is being dominated by + M
effect.
(ii) Acyl Chlorides : (r), the – I effect of Cl is more effective than + M effect, thereby
causing an opposite shift (to higher frequency).
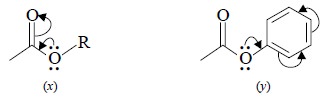
(iii) Alkyl Esters : (x), it has been observed that a conflict between I and M effects
invariably takes place in the case of esters. Here, the non-bonding electrons
residing on oxygen enhance the + M conjugation thereby decreasing the C = O
frequency.
(iv) Aryl Esters : (y), here the non-bonding electrons located on oxygen are partially
drawn into the benzene ring and thereafter their conjugation with C = O is
minimised. The net effect would be that – I effect of oxygen becomes dominant
and consequently C = O moves to a higher frequency.
(d) Field Effects : It has been observed
that two functional groups often influence each other’s vibra-tional
frequencies by a through-space interaction that may be either steric and/or
electrostatic in nature.
A typical example of ortho-chlorobenzoic
acid esters is shown below :
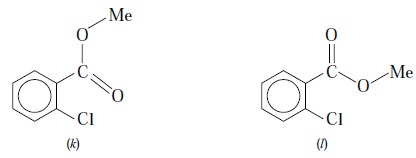
In the above instance, the field effect shifts the C = O
frequency in the rotational isomer (k)
and not in the isomer (1). As both isomers are usually found to be present
together, therefore, two C = O str. absorptions are observed in the spectrum of
this compound.
The various aspects discussed above in Sections, give a
sufficient in-depth knowledge of theoretical considerations related to the
better understanding of infrared spectroscopy.
Related Topics