Chapter: Biotechnology Applying the Genetic Revolution: Viral and Prion Infections
Infectious Prion Disease
INFECTIOUS
PRION DISEASE
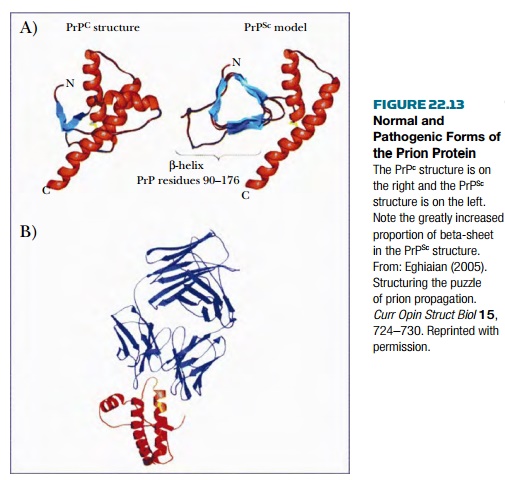
Prions are proteins with unique properties that are capable of causing inherited, spontaneous, or infectious disease. The prion protein (PrP) exists in two conformations, the normal harmless or “cellular” form ( PrP c ) and the pathogenic ( PrP Sc ) form, named after scrapie , a disease of sheep ( Fig. 22.13 ). Rogue prion proteins bind to their normal relatives and induce them to refold into the disease-causing conformation. Thus, a small number of misfolded prions will eventually subvert the population of normal proteins. Over time, this leads to neural degeneration and eventually death. Mutations within the Prnp gene that encodes the prion protein may result in prions with a greatly increased likelihood of misfolding. This causes hereditary prion disease. Several clinically different variants are known, depending on the precise location of the mutation within the prion protein and the nature of the amino acid alteration. The most common is Creutzfeldt-Jakob disease (CJD). Even normal prions occasionally misfold. The result is spontaneous prion disease, which occurs at a rate of about one per million of the human population.
If misfolded prions are transmitted to another susceptible host, the result is infectious prion disease, also known as transmissible spongiform encephalopathy (TSE) . Such an infection can be passed from one cell to another and one animal to another by entry of the PrP Sc form of the prion. The two individuals may be of the same or different species. Infection of a new victim by prions is relatively difficult. It requires uptake of rogue prion proteins from infected nervous tissue, especially brain, but the details of infection remain obscure. The best known infectious prion diseases are:
1 . Scrapie, a disease of sheep and goats
2 . Kuru , a disease of cannibals
3 . Mad cow disease , officially known as
bovine
spongiform encephalopathy
(BSE)
4 . Chronic wasting disease (CWD) of deer
and elk.
This can apparently be
transmitted via saliva, unlike the other TSEs. Scrapie is a disease of sheep
and related animals that has been recorded going back several hundred years in
Europe. The name comes from the behavior of infected sheep that constantly
scrape themselves against fences, trees, or walls and often seriously injure themselves.
Only certain breeds of sheep are susceptible, because of the slight differences
in prion sequence between breeds. Dead and decomposing sheep may contaminate
the grass of their fields with prion proteins. These are unusually stable and
long-lived and may be eaten by healthy sheep.
Kuru was transmitted by ritual
cannibalism and used to be endemic among the Fore tribe of New Guinea. The
women had the honor of preparing the brains of dead relatives and participating
in their ritual consumption. As a result, 90% of the victims were women together
with younger children who accompanied them. It may take 10 to 20 years for
symptoms to develop, but once they do, the progression from headaches to
difficulty walking to death from neural degeneration took from 1 to 2 years. No
one born since 1959, when cannibalism stopped, has developed kuru.
Brain degeneration, or
spongiform encephalopathy, due to misfolded prions is possible in any mammalian
species. In addition to scrapie, BSE, and CWD, a variety of less
well-characterized prion diseases are known in other animals. In a way, these are
really all the same disease, because there is a single prion gene encoding a
single prion protein that is found in the brain of all mammals. Symptoms vary
slightly from species to species, but after a long incubation period, the
result is degeneration and death of cells of the central nervous system. As the
popular name mad cow disease indicates, progressive degeneration of the
brain and nervous system causes the infected animals to behave bizarrely during
later stages of the disease.
Mad cow disease was spread
by overly intensive farming practices. Animal remains, including the brains,
were ground up and incorporated into animal feed. Because sheep remains were included
in feed for cows, the epidemic of mad cow disease, which began in England in
1986, was originally blamed on sheep with scrapie. However, people in England
and other European countries have eaten sheep with scrapie since the 1700s
without any noticeable ill effects. Nor have any other domestic animals,
including cows, ever caught scrapie, despite sharing the same fields. Moreover,
sheep prions are not infectious for cows. It is now thought that a random flip-flop
event converted a normal prion into the rogue form inside a cow’s brain
somewhere in England in the late 1970s or early 1980s. The rogue cow prions
were recycled in animal feed and spread, eventually causing an epidemic. After
mad cow disease broke out in England, the recycling of animal remains in feed
was prohibited and infected herds were destroyed. Mad cow disease can be
transmitted to humans, but the rate of infection is extremely low.
The first human cases were
confirmed in 1996 and were named variant CJD in an attempt to obscure their
origin. However, when the rogue cow prion infects humans, the misfolded prions
are characteristic of mad cow disease, not genuine CJD. In humans with CJD or
kuru, the precise
conformation of the misfolded prions is different. The human victims of madcow
disease are scattered randomly throughout the population, suggesting that
relativelyfew humans are actually susceptible to infection. As of 2006, about
200 people, mostly inEngland, have come down with BSE. Calculations based on
the history and age distributionof BSE in humans since the outbreak started
suggest an average incubation period of about15 years and that the number of
cases will eventually reach around 300. These estimates aremuch lower than many
earlier and rather emotional predictions and reflect the extremelylow
infectivity of prions when crossing from one species to another.
Related Topics