Chapter: Medical Immunology: Immunohematology
Immune Hemolytic Anemias
IMMUNE HEMOLYTIC ANEMIAS
The designation of hemolytic anemias includes a heterogeneous group of diseases whose common denominator is the exaggerated destruction of red cells (hemolysis).
Case 22.2
A 65-year-old man being treated for essential hypertension with a combination of thiazide and α-methyldopa was seen by his internist. He was complaining of tiredness and short-ness of breath. The following laboratory results were obtained:
Hemoglobin 10 gm/dL
Hematocrit 31%
Reticulocytes 8%
Bilirubin, direct 1.5 mg/dL
Bilirubin, total 3.6 mg/dL
Direct antiglobulin test Positive with anti-IgG
Indirect antiglobulin test Positive
Panels performed on both the serum and an eluate from the patient’s red cells revealed positive reactions with all cells tested, indicating the presence of an antibody of broad specificity.
This case raises several questions:
1. What are the two most probable causes of this patient’s anemia, and why is it im-portant to distinguish between the two?
2. What is the pathogenesis of the two types of hemolytic anemia most likely in-volved?
3. What immediate measure(s) should be instituted?
A. Autoimmune Hemolytic Anemia (Warm Antibody Type)
This is the most common form of autoimmune hemolytic anemia. It can be idiopathic (of-ten following overt or subclinical viral infection) or secondary, as shown in Table 22.5.

1. Pathogenesis
“Warm” autoimmune hemolytic anemia (AIHA) results from the spontaneous emergence of IgG antibodies, which may have a simple Rh specificity such as anti-e, or uncharacter-ized specificities common to almost all normal red cells (“public” antigens, thought to be the core of the Rh substance). In many patients one can find antibodies of more than one specificity. The end result is that the serum from patients with autoimmune hemolytic ane-mia of the warm type is likely to react with most, if not all, of the red cells tested. These an-tibodies usually cause shortening of red cell life due to the uptake and destruction by phago-cytic cells in the spleen and liver.
2. Diagnosis
Diagnosis relies on the demonstration of antibodies coating the red cells or circulating in the serum. RBC-fixed antibodies are detected by the direct antiglobulin Coombs’ test.
The test can be done using anti-IgG antiglobulin, anticomplement, or polyspecific antiglobulin serum that has both anti-IgG and anticomplement. The polyspecific or broad-spectrum antiglobulin sera produce positive results in higher numbers of patients, as shown in Table 22.6.

The search for antibodies in serum is carried out by the indirect antiglobulin test. Cir-culating antibodies are only present when the red cells have been maximally coated, and the test is positive in only 40% of the cases tested with untreated red cells. A higher posi-tivity rate (up to 80%) can be achieved by using red cells treated with enzymes such as trypsin, papain, ficin, and bromelin in the agglutination assays. The treatment of red cells with these enzymes increases their agglutinability by either increasing the exposure of anti-genic determinants or by reducing the surface charge of the red cells. In the investigation of warm-type AIHA, all tests are carried out at 37°C.
B. Cold Agglutinin Disease and Cold Agglutinin Syndrome
These diseases can also be idiopathic or secondary.
1. Pathogenesis
The cold agglutinins are classically IgM antibodies (very rarely IgA or IgG) and react with red cells at temperatures below normal body temperature.
In chronic, idiopathic, cold agglutinin diseases, the antibodies are in the vast major-ity of cases of the IgMk isotype and react with the I antigen. This is a public antigen in adults. The fetus expresses the “i” antigen, common to primates and other mammalians, which is the precursor of the “I” specificity. The newborn expresses i predominantly over I; in the adult, the situation is reversed.
In postinfectious cold agglutinin syndrome, the antibodies are also predominantly IgM but contain both k and λ light chains, suggesting their polyclonal origin. The cold ag-glutinins that appear in patients with Mycoplasma pneumoniae infections are usually reac-tive with the I antigen, while those that appear in association with infectious mononucleo-sis usually react with the i antigen.
The range of thermal reactivity of cold agglutinins may reach up to 35°C. Such tem-peratures are not difficult to experience in exposed parts of the body during the winter. Cold-induced intravascular agglutination, causing ischemia of cold-exposed areas, and hemolysis are the main pathogenic mechanisms in cold agglutinin disease.
2. Clinical Presentation
Hemolysis is usually mild but in some cases may be severe, leading to acute tubular necrosis. But in most cases the clinical picture is dominated by symptoms of cold sensitivity (Raynaud’s phenomenon, vascular purpura, and tissue necrosis in exposed extremities).
3. Laboratory Diagnosis
Testing for cold agglutinins is usually done by incubating a series of dilutions of the pa-tient’s serum (obtained by clotting and centrifuging the blood at 37°C immediately after drawing) with normal group O RBCs at 4°C. Titers up to the thousands or even millions can be observed in patients with cold agglutinin disease. Intermediate titers ( <1000) can be found in patients with Mycoplasma pneumoniae infections (postinfectious cold agglu-tinins). Low titers ( <64) can be found in normal asymptomatic individuals.
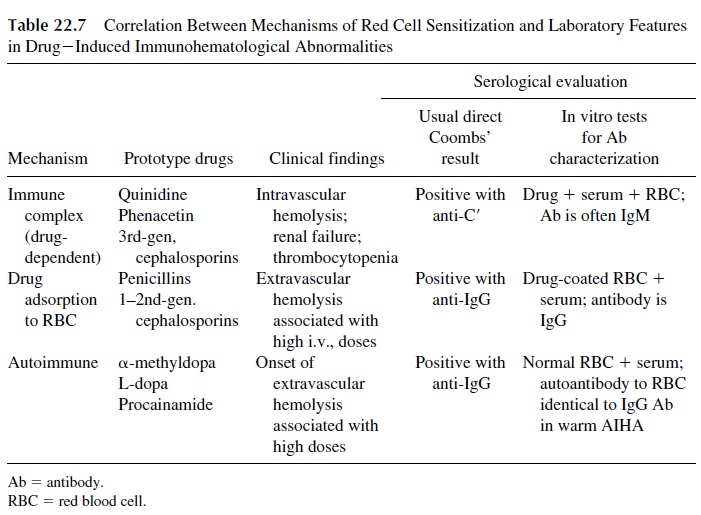
C. Drug-Induced Hemolytic Anemia
Three different types of immune mechanisms may play a role in drug-induced hemolytic anemias, as summarized in Table 22.7. It is important to differentiate between drug-in-duced hemolytic anemia and warm autoimmune hemolytic anemia, because cessation of the drug alone will usually halt the drug-induced hemolytic process.
1. Immune Complex Mechanism (Drug-Dependent Antibody Mechanism)
Traditionally this mechanism has been thought to be a part of the formation of soluble im-mune complexes between the drug and the corresponding antibodies, which is followed by nonspecific adsorption to red cells and complement activation. Alternatively, the neoanti-gen concept proposes that the drug binds transiently with the red cell, forming a “nonself” epitope, which stimulates antibody formation. The distinction between this mechanism and the drug adsorption mechanism, where a stable bond is formed between the drug and the cell membrane, may be more apparent than real. When IgM antibodies are predominantly involved, intravascular hemolysis is frequent, and the direct Coombs’ test is usually posi-tive if anticomplement antibodies are used. IgG antibodies can also form immune com-plexes with different types of antigens and be adsorbed onto red cells and platelets. In vitro such adsorption is not followed by hemolysis or by phagocytosis of red cells, but in vivo it has been reported to be associated with intravascular hemolysis.
The absorption of IgG-containing immune complexes to platelets is also the cause of drug-induced thrombocytopenia. Quinine, quinidine, digitoxin, gold, meprobamate, chlorothiazide, rifampin, and the sulfonamides have been reported to cause this type of drug-induced thrombocytopenia.
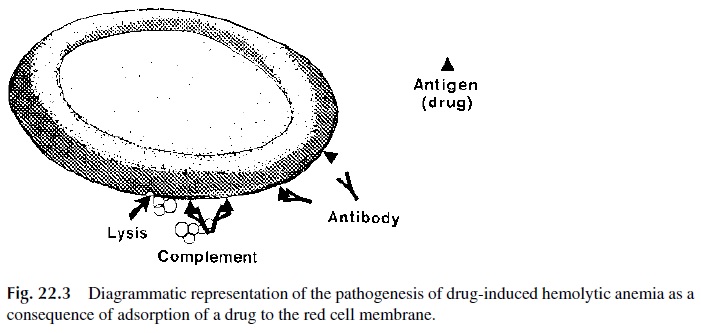
2. Drug Adsorption Mechanism
This mechanism proposes that the adsorbed drug functions as hapten and the RBC as car-rier, and an immune response against the drug ensues. The antibodies, usually IgG, are pre-sent in high titers and may activate complement after binding to the drug adsorbed to the red cells, inducing hemolysis (Fig. 22.3) or phagocytosis. Penicillin (when administered in high doses by the intravenous route) and cephalosporins can induce this type of hemolytic anemia.
Some cephalosporins (such as cephalothin) also have been shown to modify the red cell membrane that becomes able to adsorb proteins nonspecifically, a fact that can lead to a positive direct Coombs’ test but not to hemolytic anemia.
3. Autoimmunity Induction Mechanism
The drug most implicated in this form of drug-induced hemolytic anemia is α-methyldopa (Aldomet). Ten to 15% of the patients receiving the drug will have a positive Coombs’ test, and 0.8% of the patients develop clinically evident hemolytic anemia. It is particularly in-teresting from the pathogenic point of view in that it is indistinguishable from a true warm autoimmune hemolytic anemia.
α-Methyldopa is unquestionably the trigger for this type of anemia, but the antibod-ies are of the IgG1 isotype and react with Rh antigens. It is believed that the drug changes the membrane of red cell precursors, causing the formation of antibodies reactive with a modified Rh precursor. Once formed, the anti–red cell antibodies will react in the absence of the drug as true autoantibodies.
Other drugs such as L-dopa, procainamide, and some nonsteroidal anti-inflammatory drugs can also act by this mechanism. Both α-methyldopa and L-dopa also stimulate the production of antinuclear antibodies.
D. Treatment
Blood transfusions may be necessary in emergency situations but are made difficult when autoantibodies to red cell antigens are widely represented in the population. The serum from a patient with cold or warm-type AIHA typically agglutinates all the red cells in an antibody identification panel. It is most important to determine if there are clinically sig-nificant underlying alloantibodies that may be masked by autoantibody. The patient’s red cells may be pretreated in a manner to enhance removal of autoantibody from the serum. After one or more autoadsorptions, the adsorbed serum may be used for alloantibody de-tection and cross-matching.
Glucocorticoids are the first line of treatment in patients with symptomatic warm AIHA. Other immunosuppressive drugs can be tried in patients not responding to gluco-corticoids. Splenectomy can be useful in individuals who do not respond to glucocorti-coids, when there is a marked predominance of red cell sequestration in the spleen. In such cases splenectomy leads to a longer half-life of the patient’s red cells.
Glucocorticoids and splenectomy are generally ineffective in treating cold agglutinin disease. Patients should be kept warm, especially their extremities. If transfusions are to be administered, an approved blood warmer should be used immediately prior to transfusion.
In cases of drug-induced hemolytic anemia the offending drug should be withdrawn and hemolysis should resolve.
Case 22.2 Revisited
2. Warm-type hemolytic anemia is an autoimmune condition, which can present itself as the only manifestation of autoimmunity or as part of the constellation of a systemic autoimmune disease. The autoantibodies are of broad specificity, reacting with public erythrocyte antigens expressed by almost every individual. Drug-induced hemolytic anemia can be caused by an-tibodies directed to the drug, due either to previous adsorption of the drug to the red cell or to adsorption of preformed antigen-antibody complexes to the red cell, or (as is the case in hemolytic anemia associated with α-methyldopa by anti–red cell antibodies identical to those detected in true autoimmune hemolytic anemia. How α-methyldopa causes the production of these antibodiesis the object of speculation. It is believed that the drug may alter the conformation of the Rh complex on the red cell membrane, triggering the synthesis of antibodies that cross-react with unchanged Rh substances. Thus, once induced, the autoantibody recognized the red cell rather than α-methyldopa, and withdrawal of the drug may not result in immediate improvement.
3.In all cases of drug-induced hemolytic anemia it is important to stop the admin- istration of the drug as soon as the diagnosis is established. In the case of α- methyldopa–induced hemolytic anemia the improvement will be gradual, be-cause the autoantibodies react with the red cells, rather than with the drug. However, the antibody titers will decrease with time, and after a point their con-centration may be still sufficient to cause a positive direct antiglobulin test but not to cause significant anemia. In contrast, treatment of true autoimmune hemolytic anemia is rather more complex, involving administration of steroids and, in cases not responding to steroids, splenectomy and/or administration of immunosuppressive drugs.
Related Topics