Chapter: Genetics and Molecular Biology: Oncogenesis, Molecular Aspects
Identifying a Nucleotide Change Causing Cancer
Identifying a Nucleotide Change Causing Cancer
As
mentioned above, since mutagens are carcinogenic, it is likely that some
cancers are caused by mutation. How could we determine the nucleotide change or
changes that cause a human cell to be trans-formed? If such a change could be
detected, it would definitively identify the altered gene. This would assist
the study of the gene product’s normal cellular function. In principle,
comparing the nucleotide se-quences of all the chromosomes from a normal cell
and a transformed cell would reveal the change. Even if such an ill-advised
attempt to sequence about 6 × 109
base pairs of DNA could be accomplished, however, the important change would
likely be masked by a multitude of extraneous random nucleotide changes.
To reduce
the problem of locating the nucleotide change that causes a cancer to
manageable proportions, a powerful selection technique for the critical DNA
fragment is required. The best selection is to use the ability of the DNA to
induce uncontrolled growth. Cells growing in culture can be used for such a
screening. For example, one line of mouse cells, NIH 3T3 cells, has been
adapted for continuous growth in culture, in contrast to primary cell lines
that grow for only a limited number of generations. Nonetheless, the NIH 3T3
cells still display density-depend-ent growth regulation and cease further growth
when a monolayer of cells has grown on the container surface. Thus, the cells
can be consid-ered to be part way along the pathway leading to transformation.
If one of these cells is transformed to a cancerous state, it loses its growth
regulation, and its descendants continue growing when the other cells in the
monolayer have stopped. The resulting pile of cells is detectable through a
microscope, and is called a focus. Weinberg and others have
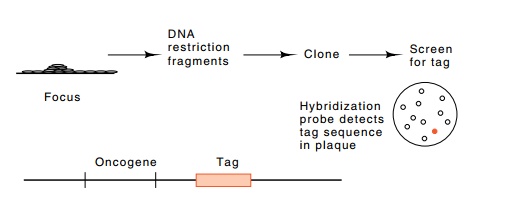
Figure
23.5 The use of a tag to permit
detection of an oncogene when cloningcrude DNA from a transformed cell into E. coli.
The desired plaque contains material complementary to the tag known to be
associated with the oncogene.
Although the focus assay for transforming DNA
fragments could be used in conjunction with a straightforward biochemical
purification of the desired DNA fragments, genetic engineering methods
permitted more rapid isolation of the DNA. The basic trick was to tag
restriction fragments of the DNA extracted from a tumor before it was used to
transform cells to grow foci. DNA extracted from cells of a focus was then
cloned in E. coli and the colonies or
plaques were screened using the tag sequence as a probe. Of course, the tag had
to be a sequence not normally found in the NIH 3T3 cells or in Escherichia coli (Fig. 23.5). Therefore,
the clone containing the human-derived sequence capable of transforming the
cells to density-independent growth could be detected.
Two methods can be used for tagging the oncogene
isolated from cancerous cells. One makes use of naturally occurring sequences.
As mentioned, in humans, a particular sequence called the Alu sequence is widely scattered throughout the genome and is
repeatedhundreds of thousands of times. Because this sequence contains two Alu restriction enzyme cleavage sites,
cleavage of human DNA with Alu restriction
enzyme generates hundreds of thousands of copies of a specific size of DNA
fragment in addition to all the other random sized fragments. The mouse genome
lacks this sequence. Simply because of its high repetition number and wide
dispersal through the human genome, one of the Alu sequences was likely to lie near the mutated gene responsible
for the uncontrolled cell growth and transformation of cells to form a focus.
Indeed, one did, and Weinberg was able to use this as a tag of the human gene
that was mutated to cause the human cancer.
A second method for tagging the oncogene is direct
and general. DNA isolated from a tumor can be digested with a restriction
enzyme, and a synthetic DNA containing the desired tagging sequence can be
ligated to it. Then the mixture of DNAs can be transformed into cultured mouse
cells for selection of the fragment containing the
oncogene. This is followed by cloning in bacteria and identification of the
oncogene clone with the use of the tag sequence as a probe.
A minor technical point must be mentioned. The
cultured cells that take up the human DNA usually take up many molecules.
Therefore, although a focus of cells will contain the desired transforming
gene, it is likely to contain a number of other irrelevant human sequences as
well. These also will contain the tagging sequence. This problem is eliminated
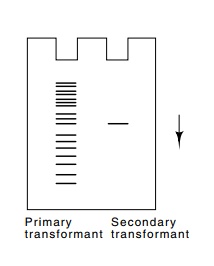
by using DNA extracted from the primary round of transfor-mation in a second
round of transformation. Indeed, after the first cycle, a Southern transfer
prepared from DNA of a transformed cell reveals many sizes of restriction
fragments containing the human-derived Alu
tagging sequence (Fig. 23.6), but after a second cycle of transformation, all
the transformants contain only a single copy of the tagging sequence.
Presumably this is directly linked to the stretch of DNA that is respon-sible
for the transformation.
The techniques described above permitted the
cloning and isolation of DNA fragments 5 to 10 Kb long that contained the
cellular oncogene from bladder carcinoma cells that could transform NIH 3T3
cells. Once these fragments were obtained, they could be used as probes in
screen-ing genomic libraries made from DNA isolated from noncancerous human
cells. Thus the normal gene predecessor of the oncogene could also be obtained.
Then the question was merely to determine the difference between the two DNAs.
Again, they could have been totally sequenced, but ingenuity reduced the work.
Restriction fragments be-tween the two were exchanged to find a small DNA
segment that was necessary for transformation (Fig. 23.7).
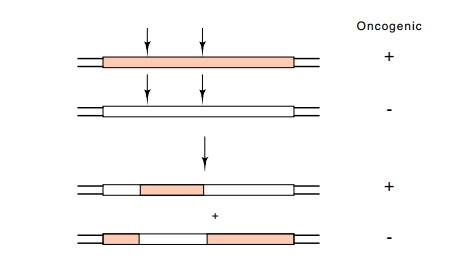
Figure 23.7 Restriction fragment interchange between the cellular homologof the oncogene and the oncogene itself, which can identify the fragment containing the critical base change.
A single nucleotide difference was found to separate the normal cellular prototype and its oncogenic variant. The most common change in the bladder cancers was found to occur in the twelfth amino acid of a protein. Since both Northern transfers of total cellular RNA and immune precipitations of the actual gene product involved show that neither transcription of the gene nor its translation efficiency is appre-ciably altered by the mutation, the structural change in the protein must be the cause of its oncogenicity, and this has been observed. The gene encodes a protein called Ras that hydrolyzes GTP to GDP. The mutant protein possesses reduced hydrolytic activity.
Related Topics