Chapter: Medical Microbiology: An Introduction to Infectious Diseases: Papovaviruses
Human Diseases Caused By Unconventional Viral Agents: Subacute Spongiform Encephalopathies
HUMAN DISEASES CAUSED BY UNCONVENTIONAL VIRAL AGENTS: SUBACUTE SPONGIFORM ENCEPHALOPATHIES
A group of progressive degenerative diseases of the CNS has been shown to be caused by infectious agents with unusual physical and chemical properties, which are now known as prions. The Nobel prize in Medicine for 1997 was awarded to Stanley Prusiner for his work in identifying the role of prions in disease. Prions cause bovine spongiform encephalopathy in cattle, scrapie in sheep, and five fatal CNS diseases in humans
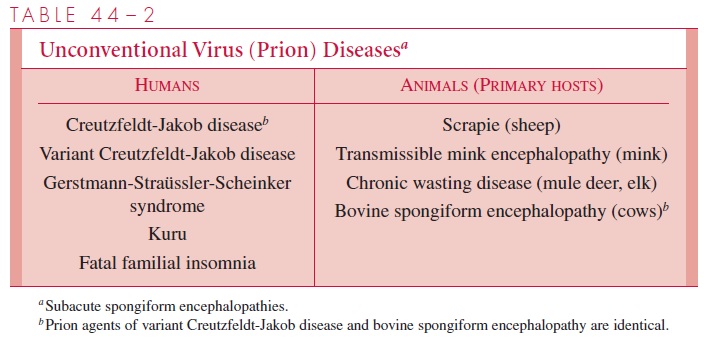
(Table 44–2). Prions can be the etiologic agents of inherited, communicable, or sporadic diseases. The pathogenesis of these illnesses is not well understood, but the pathologic and clinical features are similar. Varying degrees of neuronal loss and astrocyte prolifera-tion occur. The diseases are known as “spongiform” encephalopathies because of the vac-uolar changes in the cortex and cerebellum. The incubation periods of these diseases are months to years, and their courses are protracted and inevitably fatal.
A prion is defined as a “small proteinaceous infectious particle” that is not inactivated by procedures that destroy nucleic acids (Table 44–3). They are small with diameters of 5–100 nm or less, produce characteristic infections, and can remain viable even in forma-linized brain tissue for many years. They are resistant to ionizing radiation, boiling, and many common disinfectants. Recognizable virions have not been found in tissues by elec-tron microscopy, and the agents have not been grown in cell culture.

A prion is composed of proteins encoded by a normal cellular gene. The protein, des-ignated PrPc, is converted from a normal benign form into a disease-causing form by a change in conformation to a protein designated PrPsc (for the scrapie protein). Brain ex-tracts from scrapie-infected animals contain PrPsc, which is not found in the brains of normal animals; PrPsc is the prion that is responsible for transmission and infection. The conformational change is also the way that prions multiply; that is, contact with PrPsc re-sults in a conformational change of the normal host cell protein PrPc and the formation of additional PrPsc. Proliferation of PrPsc prions and the consequent pathology results from this process. During scrapie infection, prion protein may aggregate into birefringent rods and form filamentous structures termed scrapie-associated fibrils (Fig 44–1), which are found in membranes of scrapie-infected brain tissues.

Kuru
Kuru was a subacute, progressive neurologic disease of the Fore people of the Eastern Highlands of New Guinea. The disease was brought to the attention of the Western world by Gadjusek and Zigas in 1957. Although the illness was localized and decreasing in inci-dence, its study has thrown light on the transmissibility and infectious nature of similar encephalopathies. Epidemiologic studies indicated that kuru usually afflicted adult women, or children of either sex. The disease was rarely observed outside of the Fore re-gion, and outsiders in the region did not contract the disease. The symptoms and signs were ataxia, hyperreflexia, and spasticity, which led to progressive dementia, starvation, and death. Pathologic examination revealed changes only in the CNS, with diffuse neu-ronal degeneration and spongiform changes of the cerebral cortex and basal ganglia. No inflammatory response was apparent. Inoculation of infectious brain tissue into primates produced a disease that caused similar neurologic symptoms and pathologic manifesta-tions after an incubation period of approximately 40 months. Epidemiologic studies indi-cated that transmission of the disease in humans was associated with ingestion of a soup made from the brains of dead relatives and eaten in honor of the deceased. Clinical dis-ease developed 4 to 20 years after exposure. Since the elimination of cannibalism from the Fore culture, kuru has disappeared.
Creutzfeldt–Jakob Disease
Creutzfeldt–Jakob disease is a progressive, fatal illness of the CNS that is seen most fre-quently in the sixth and seventh decades of life. The initial clinical manifestations are a change in cerebral function, usually diagnosed initially as a psychiatric disorder. Forget-fulness and disorientation progress to overt dementia and the development of changes in gait, increased tone in the limbs, involuntary movement, and seizures. These manifesta-tions resemble those of kuru. The disorder usually runs a course of 4–7 months, eventu-ally leading to paralysis, wasting, pneumonia, and death.
Creutzfeldt–Jakob disease is found worldwide, with an incidence of disease of one case per million per year. The mode of acquisition is unknown, but it occurs both sporadi-cally (85%) and in a familial pattern (15%). Infection has also been transmitted by dura mater grafts, corneal transplants, by contact with contaminated electrodes or instruments used in neurosurgical procedures, and by pituitary-derived human growth hormone. The latter was responsible for more than 100 cases. The incubation period of the disease is ap-proximately 3 to greater than 20 years.
The pathology of Creutzfeldt–Jakob disease is identical to that of kuru. It has been transmitted to chimpanzees, mice, and guinea pigs by inoculation of infected brain tissue, leukocytes, and certain organs. High levels of infectious agent have been found, espe-cially in the brain, where they may reach 10 7 infectious doses per gram of brain tissue. Nonpercutaneous transmission of disease has not been observed, and there is no evidence of transmission by direct contact or airborne spread.
Brains from patients with Creutzfeldt–Jakob disease have the birefringent rods and fibrillar structures noted in scrapie (see Fig 44–1). Identification of PrPsc and antibodies di-rected against it may become a useful diagnostic adjunct to neuropathologic examination of brain tissue. Pathologic examination of brain tissue is the only definitive diagnostic test.
There is no effective therapy for Creutzfeldt–Jakob disease, and all cases have been fatal. The small risk of nosocomial infection is related only to direct contact with infected tissue. Stereotactic neurosurgical equipment, especially that used in patients with undiag-nosed dementia, should not be reused. In addition, organs from patients with undiagnosed neurologic disease should not be used for transplants. Growth hormone from human tis-sue has now been replaced by a recombinant genetically engineered product. The agent of Creutzfeldt–Jakob disease has not been transmitted to animals by inoculation of body se-cretions, and no increased risk of disease has been noted in family members or medical personnel caring for patients. Recommendations for disinfection of potentially infectious material include treatment for 1 hour with 2 N NaOH or by autoclaving at 132°C for 60–90 minutes. Others recommend even more extensive treatment such as combining these two procedures to ensure inactivation.
Gerstmann-Straüssler-Scheinker Disease
Gerstmann-Straüssler-Scheinker disease is similar to Creutzfeldt–Jakob disease but occurs at a younger age (fourth to fifth decade). Cerebellar ataxia and paralysis are common, but dementia is less often seen. The disease evolves over several years. It was originally thought to be familial but also occurs sporadically, very rarely.
Fatal Familial Insomnia
This is a recently recognized familial prion disease in which a syndrome of sleeping diffi- culty is followed by progressive dementia. It occurs in patients aged 35 to 61, culminating in death within 13 to 25 months. The infectious agent has been transmitted to experimen- tal animals.
Bovine Spongiform Encephalopathy (“Mad Cow Disease”) and “Variant Creutzfeldt–Jakob Disease”
Bovine spongiform encephalopathy (BSE) was identified in 1986, after it began striking cows in the United Kingdom, causing them to become uncoordinated and unusually ap-prehensive. The source of the emerging epidemic was soon traced to a food supplement that included meat and bone meal from dead sheep. The methods for processing sheep carcasses had been changed in the late 1970s. Once they would have eliminated the “scrapie agent” in the supplement, but now they apparently did not.
To combat BSE, the British government banned the use of animal-derived feed sup-plements in 1988, and the epidemic among cattle, which peaked at nearly 40,000 cases in 1992, decreased to less than 4000 new cases in 1997. By February 2002, most European countries had reported cases of BSE but new infections have largely ceased as a result of imposing tight controls on cattle feed. The United States has been spared, as measured by over 19,000 cattle brain examinations. The incubation period in cattle was determined to be 2 to 8 years. In addition to the incoordination and apprehension, the cows exhibited hyperesthesia, hyperreflexia, muscle fasciculations, tremors, and weight loss. Autonomic dysfunction was frequently manifested as reduced rumination, bradycardia and other car-diac arrhythmias.
Unfortunately, the prion that causes BSE survived the heat of cooking and was trans-mitted to humans who inadvertently consumed infected bovine neural tissue or bone marrow (both are sometimes found in processed meats, depending on the rendering pro-cedures used). To date, over 100 humans with “variant Creutzfeldt–Jakob disease,” have died in the United Kingdom. The cases frequently present in young adults as psychiatric problems progressing to neurologic changes and dementia, with death in an average of 14 months. It appears that destruction of diseased cattle and the changes in livestock feeds have prevented further cases.
Related Topics