Chapter: Essential Microbiology: Antimicrobial Agents
How do antibiotics work?
How do antibiotics work?
All antibiotics have the common property of interfering in some way with a normal, critical function of the target bacterial cell. The most commonly used antibiotics exert their effect by one of the following methods:
1 Inhibition of cell wall synthesis (group I)
2 Disruption of cell membranes (group II)
3 Interference with protein synthesis (group III)
4 Interference with nucleic acid synthesis (group IV)
Table 14.2 lists examples of each group. Those antibiotics belonging to groups I and III are better able to discriminate between procaryotic and eucaryotic cells, and consequently show more selective toxicity and a higher therapeutic index.
Group I: Inhibitors of cell wall synthesis
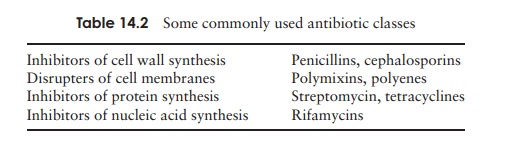
The main group which work in this way are the β-lactam antibiotics, so-called because they contain a β-lactam ring in their structure. Included among this group are the penicillins and the cephalosporins.
You may recall from our discussion of bacterial cell wall structure that an important factor in the strengthening of the peptidoglycan component of the bacterial cell walls is the cross-linking of chains by transpeptidation. It is this process which is acted on by the β-lactams; they bind irreversibly to the transpeptidase enzyme, forming covalent bonds with a serine residue within the enzyme’s active site. The cell wall continues to form, but becomes progressively weaker as more new, unlinked, peptidoglycan is set down. Since bacteria are generally to be found in a hypotonic environment, as the wall weakens, water enters the cell, leading to swelling and then lysis.
Penicillins
The firstβ-lactam antibiotic to be discovered was benzylpenicillin, orpenicillin-G, whose action is restricted to Gram-positive bacteria, because it is unable to penetrate the Gram-negative cell wall. It is effective against Gram-positive bacteria when administered intramuscularly, but cannot be taken by mouth because it is broken down in the acid conditions of the stomach. Another naturally occurring penicillin, penicillin-V, represented an advance inasmuch as it is less acid-labile and can therefore be taken orally. All the penicillins are based on a core structure or nucleus called 6-amino-penicillanic acid (Figure 14.2); extensive research has led to the develop-ment of many variants of this, the so-called semisyntheticpenicillins. These have attached to their nucleus novelside chains not encountered in nature, and have over-come some of the problems inherent in naturally occur-ring penicillins such as instability and narrow specificity (Figure 14.3).
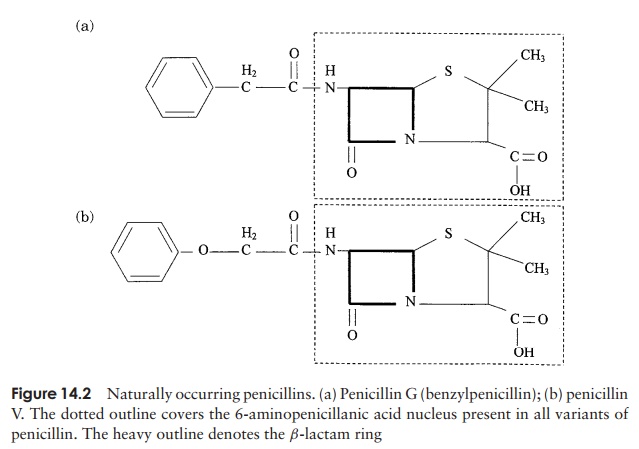
Ampicillin is a semi-synthetic penicillin that has a broader specificity than Penicillin G; it is appreciably more effective against Gram-negative bacteria such as Salmonella and E. coli, its hydrophobic nature making it better able to penetrate theirouter membrane. It has the additional benefit of being acid-stable and can therefore be taken orally.
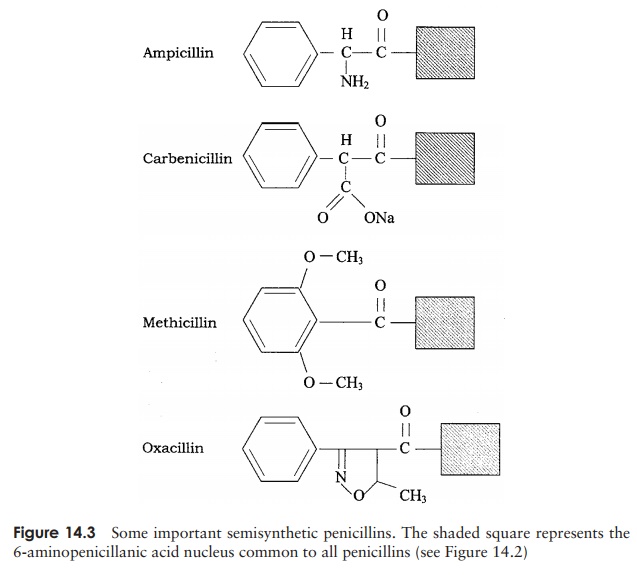
Another drawback to natural penicillins is that they are susceptible to naturally occurring bacterial β-lactamases (also called penicillinases), which breaks a bond in the β-lactam core of the penicillin molecule (Figure 14.4). Sometimes, β-lactam antibiotics
are taken in combination with a β-lactamase inhibitor such as clavulanic acid. This binds to the β-lactamase with a high affinity, preventing it from acting on the antibiotic. Some semisynthetic penicillins such as methicillin and oxacillin are resistant to attack by the β-lactamases that can render certain bacteria resistant to their naturally occurring forms.
Penicillin is not an appropriate treatment for the estimated 1–5 per cent of adults who show an allergic reaction to it; in extreme cases, death from anaphylacticshock can result.
Cephalosporins
The cephalosporins, like the penicillins, have a structure based on aβ-lactam ring (Figure 14.5). They also exert their effect on transpeptidases, but gen-erally have a broader specificity and are more resistant to the action of β-lactamases. Ceftriaxone, for example, is now used in the treatment of gonorrhoeal infections, caused by penicillin-resistant strains of Neisseria gonorrhoeae. In addition, patients who are allergic to penicillin are often treated with cephalosporins. Cephalosporins were first
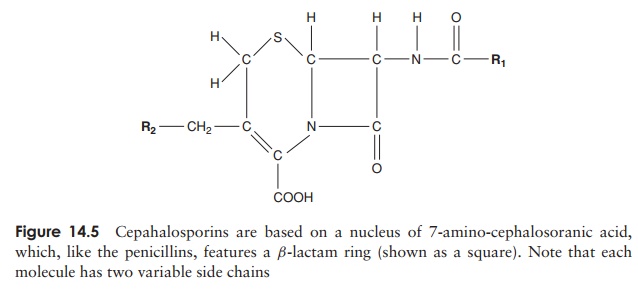
isolated in the late 1940s from a marine fungus called Cephalosporium acremonium, and came into general use in the 1960s. So-called second, third and fourth genera-tion cephalosporins have been developed to widen the spectrum of activity to include many Gram-negative organisms, and to keep one step ahead of pathogens developing resistance to earlier versions.
Both penicillins and cephalosporins are also used prophylactically, that is, in the prevention of infections, prior to surgery in particularly vulnerable patients.
Other antibiotics that affect the cell wall
Carbapenems areβ-lactam antibiotics pro-duced naturally by a species of Streptomyces. A semisynthetic form, imipenem, is active against a wide range of Gram-positive and -negative bacteria, and is used when resis-tance to other β-lactams has developed.
Bacitracin and vancomycin are two other antibiotics that exert their effect on the cell wall, but by a different mechanism. Bacitracin is derived from species of Bacillus and acts on bactoprenol pyrophosphate, the lipid carrier molecule responsible for transport-ing units of peptidoglycan across the cell membrane to their site of incorporation into the cell wall. Its use is restricted to topical (surface) application, since its use internally can cause kidney damage. Vancomycin is a highly toxic antibiotic with a narrow spectrum of use against Gram-positive organisms such as streptococci and staphylococci. It is particularly important in its use against infections caused by organ-isms resistant to methicillin and the cephalosporins, such as methicillin-resistant Staphy-lococcus aureus (MRSA) (see Resistance to Antibiotics below). It is not absorbed fromthe gastrointestinal tract and is therefore most commonly administered intravenously.
Group II: Antibiotics that disrupt cell membranes
Polymixins are a class of antibiotic that act by disrupting the phospholipids of the cytoplasmic membrane and causing leakage of cell contents. Produced naturally by a species of Bacillus, polymixins are effective against pseudomonad infections of wounds and burns, often in combination with bacitracin and neomycin (an inhibitor of protein synthesis; see below). Their toxicity makes themunsuitable for internal use.
Group III: Inhibitors of protein synthesis
Antibiotics that act by affecting protein synthesis generally have a relatively broad spec-trum of action. As we saw in our historical review earlier, streptomycin was the first antibiotic that was shown to be effective against Gram-negative organisms. Its discovery in 1943 was particularly welcome since such organisms were unaffected by penicillin or sulphonamides. It proved to be particularly useful in the treatment of tuberculosis, the causative agent of which, Mycobacterium tuberculosis, is protected against the effects of penicillin by the waxy layer of mycolic acids in its cell wall.
Streptomycin belongs to a group of antibiotics called aminoglycosides, which act by binding to the 30S subunit of the bacterial ribosome, preventing attachment of the 50S subunit to the initiation complex (Figure 14.6). They can thus discriminate between procaryotic (70S) and eucaryotic (80S) ribosomes, and consequently have a relatively high therapeutic index (although not as high as cell wall inhibitors). Other members of this group are gentamicin, kanamycin and neomycin. Like some other ‘wonder drugs’, streptomycin has proved to have undesirable side-effects; these have led to it being replaced in most applications by safer alternatives. In addition, bacterial resistance to streptomycin is widespread, further diminishing its usefulness. Use of the aminoglycosides as a group has diminished since the development of later generation cephalosporins and the tetracyclines.
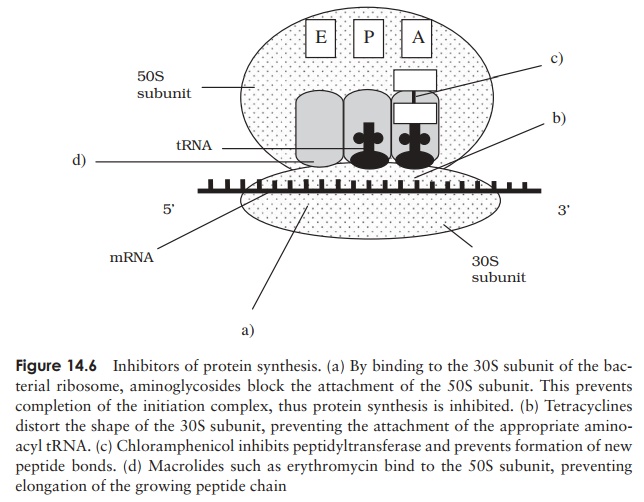
Tetracyclines also work by binding to the 30S ribosomal subunit, preventing theattachment of aminoacyl tRNA, and therefore extension of the peptide chain (Fig-ure 14.6). They are yet another group of antibiotics produced by Streptomyces spp. Both natural and semisynthetic tetracyclines are easily absorbed from the intestine, allowing them to be taken orally. Coupled with their broad specificity (the broadest of any antibiotic), this led to inappropriately widespread use in the years following their discovery, sometimes resulting in complications caused by the destruction of the normal resident microflora. Tetracyclines are still used for a number of applications, notably to treat a variety of sexually transmitted diseases.
Two important antibiotics which act on the larger, 50S, subunit of the procaryotic ribosome are erythromycin and chloramphenicol. Both combine with the subunit in such a way as to prevent the assembly of amino acids into a chain (Figure 14.6). Chlo-ramphenicol was the first antibiotic to be discovered with a broad spectrum of activity; it also derives originally from Streptomyces spp., but is nowadays produced synthet-ically. Its use has become severely restricted since it was shown to have some serious side-effects, notably on the bone marrow, but it remains the agent of choice for the treatment of typhoid fever.
Erythromycin is the best known of the macrolide group of antibiotics. Unlike chlo-ramphenicol, it has a large hydrophobic molecule and is unable to gain access to most Gram-negative bacteria, thus restricting its spectrum of activity. Erythromycin can be taken orally and has a similar spectrum of activity to penicillin G; it is often used instead of penicillin in the treatment of staphylococcal and streptococcal infections in children. It is particularly appropriate for this application as it is one of the least toxic of all commonly used antibiotics.
Group IV: Inhibitors of nucleic acid synthesis
Rifampin belongs to a group of agents called rifamycins. It acts by inhibiting the enzyme RNA polymerase, thereby preventing the production of mRNA. Rifampin is used against the mycobacteria that cause tuberculosis, an application for which its ability to penetrate tissues makes it well suited. Unlike most antibiotics, rifampin interacts with other drugs, often reducing or nullifying their effect. When used in high doses, it has the unusual side-effect of turning secretions such as tears, sweat and saliva, as well as the skin, an orange-red colour. As we have already seen, the quinolone group of synthetic antimicrobial drugs act by disrupting DNA replication.
Related Topics