Chapter: Basic & Clinical Pharmacology : Aminoglycosides & Spectinomycin
General Properties of Aminoglycosides
AMINOGLYCOSIDES
The aminoglycosides
include streptomycin, neomycin,
kanamy-cin, amikacin, gentamicin, tobramycin, sisomicin, netilmicin, and
others. They are used most widely in combination with a β-lactam antibiotic in
serious infections with gram-negative bac-teria, in combination with vancomycin
or a β-lactam
antibiotic for gram-positive endocarditis, and for treatment of tuberculosis.
General Properties of
Aminoglycosides
A. Physical and Chemical Properties
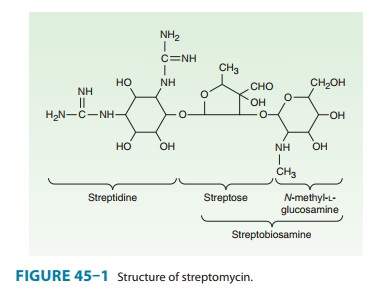
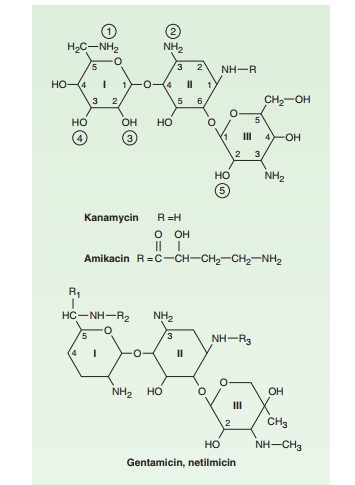
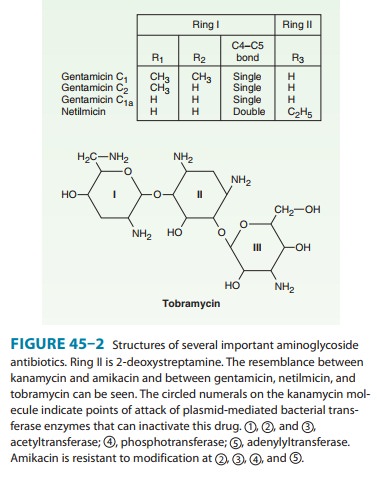
Aminoglycosides
have a hexose ring, either streptidine (in streptomy-cin) or 2-deoxystreptamine
(in other aminoglycosides), to whichvarious amino sugars are attached by
glycosidic linkages (Figures 45–1 and 45–2). They are water-soluble, stable in
solu-tion, and more active at alkaline than at acid pH.
B. Mechanism of Action
The mode of action of
streptomycin has been studied far more closely than that of other
aminoglycosides, but they probably all act similarly. Aminoglycosides are
irreversible inhibitors of protein synthesis, but the precise mechanism for
bactericidal activity is not known. The initial event is passive diffusion via
porin channels across the outer membrane (see Figure 43–3). Drug is then
actively transported across the cell membrane into the cytoplasm by an
oxygen-dependent process. The transmembrane electro-chemical gradient supplies
the energy for this process, and trans-port is coupled to a proton pump. Low
extracellular pH and anaerobic conditions inhibit transport by reducing the
gradient. Transport may be enhanced by cell wall-active drugs such as
peni-cillin or vancomycin; this enhancement may be the basis of the synergism
of these antibiotics with aminoglycosides.
Inside
the cell, aminoglycosides bind to specific 30S-subunit ribosomal proteins (S12
in the case of streptomycin). Protein synthesis is inhibited by aminoglycosides
in at least three ways (Figure 45–3): (1) interference with the initiation
complex of peptide
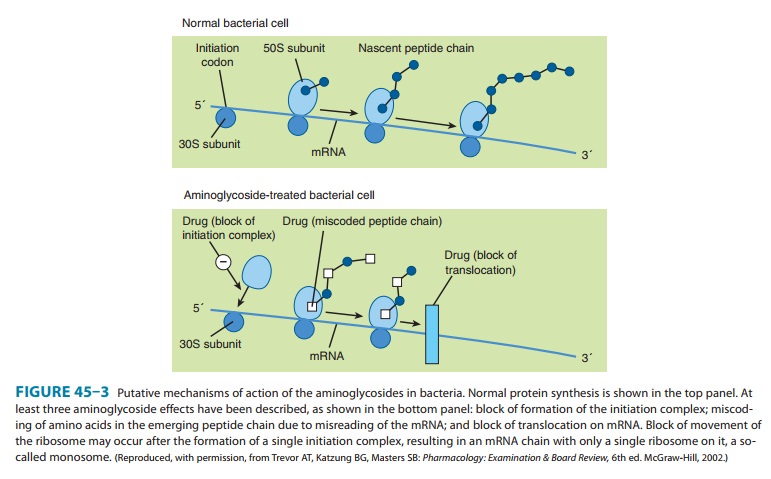
formation;
(2) misreading of mRNA, which causes incorporation of incorrect amino acids
into the peptide and results in a nonfunctional or toxic protein; and (3)
breakup of polysomes into nonfunctional monosomes. These activities occur more
or less simultaneously, and the overall effect is irreversible and lethal for
the cell.
C. Mechanisms of Resistance
Three principal
mechanisms have been established: (1) production of a transferase enzyme or
enzymes inactivates the aminoglycoside by adenylylation, acetylation, or
phosphorylation. This is the prin-cipal type of resistance encountered
clinically. (Specific transferase enzymes are discussed below.) (2) There is
impaired entry of aminoglycoside into the cell. This may be genotypic,
resulting from mutation or deletion of a porin protein or proteins involved in
transport and maintenance of the electrochemical gradient; or phenotypic, eg,
resulting from growth conditions under which the oxygen-dependent transport
process described above is not func-tional. (3) The receptor protein on the 30S
ribosomal subunit may be deleted or altered as a result of a mutation.
D. Pharmacokinetics and Once-Daily Dosing
Aminoglycosides are absorbed very poorly from the intact gastro-intestinal tract, and almost the entire oral dose is excreted in feces after oral administration. However, the drugs may be absorbed if ulcerations are present. After intramuscular injection, aminoglyco-sides are well absorbed, giving peak concentrations in blood within 30–90 minutes. Aminoglycosides are usually administered intravenously as a 30- to 60-minute infusion; after a brief distribu-tion phase, this results in serum concentrations that are identical with those following intramuscular injection. The normal half-life of aminoglycosides in serum is 2–3 hours, increasing to 24–48 hours in patients with significant impairment of renal function. Aminoglycosides are only partially and irregularly removed by hemodialysis—eg, 40–60% for gentamicin—and even less effec-tively by peritoneal dialysis. Aminoglycosides are highly polar compounds that do not enter cells readily. They are largely excluded from the central nervous system and the eye.
In the effect of each
individual drug, ie, the killing effect of the combina-tion is more than
additive. presence of active inflammation, however, cerebrospinal fluid levels
reach 20% of plasma levels, and in neonatal meningitis, the levels may be
higher. Intrathecal or intraventricular injection is required for high levels
in cerebrospinal fluid. Even after parenteral admin-istration, concentrations
of aminoglycosides are not high in most tissues except the renal cortex.
Concentration in most secretions is also modest; in the bile, it may reach 30%
of the blood level. With prolonged therapy, diffusion into pleural or synovial
fluid may result in concentrations 50–90% of that of plasma.
Traditionally,
aminoglycosides have been administered in two or three equally divided doses
per day in patients with normal renal function. However, administration of the
entire daily dose in a single injection may be preferred in many clinical
situations. Aminoglycosides have concentration-dependent
killing; that is, increasing concentrations kill an increasing proportion
of bacteria and at a more rapid rate. They also have a significant postantibioticeffect, such that the
antibacterial activity persists beyond the timeduring which measurable drug is
present. The postantibiotic effect of aminoglycosides can last several hours.
Because of these proper-ties, a given total amount of aminoglycoside may have
better efficacy when administered as a single large dose than when administered
as multiple smaller doses. When administered with a cell wall-active antibiotic
(a β
lactam or vancomycin), aminogly-cosides exhibit synergistic killing against certain bacteria. The effect of the
drugs in combination is greater than the anticipated
Adverse effects from
aminoglycosides are both time- and concentration-dependent. Toxicity is
unlikely to occur until a certain threshold concentration is reached, but once
that concen-tration is achieved, the time beyond this threshold becomes
criti-cal. This threshold is not precisely defined, but a trough concentration
above 2 mcg/mL is predictive of toxicity. At clini-cally relevant doses, the
total time above this threshold is greater with multiple smaller doses of drug
than with a single large dose.
Numerous clinical
studies demonstrate that a single daily dose of aminoglycoside is just as
effective—and probably less toxic— than multiple smaller doses. Therefore, many
authorities now recommend that aminoglycosides be administered as a single
daily dose in many clinical situations. However, the efficacy of once-daily
aminoglycoside dosing in combination therapy of enterococ-cal and
staphylococcal endocarditis remains to be defined, and the standard low-dose,
thrice-daily administration is still recom-mended. In contrast, there are
limited data supporting once-daily dosing in streptococcal endocarditis. The
role of once-daily dosing in pregnancy and in neonates also is not well
defined.
Once-daily dosing has
potential practical advantages. For example, repeated determinations of serum
concentrations are probably unnecessary unless aminoglycoside is given for more
than 3 days. A drug administered once a day rather than three times a day is
less labor intensive. And once-a-day dosing is more feasible for outpatient
therapy.
Aminoglycosides are
cleared by the kidney, and excretion is directly proportional to creatinine
clearance. To avoid accumulation and toxic levels, once-daily dosing of
aminoglycosides is generally avoided if renal function is impaired. Rapidly
changing renal function, which may occur with acute kidney injury, must also be
monitored to avoid overdosing or underdosing. Provided these pitfalls are
avoided, once-daily aminoglycoside dosing is safe and effective. If the
creatinine clearance is > 60 mL/min, then a single daily dose of 5–7 mg/kg
of gentamicin or tobramycin is recom-mended (15 mg/kg for amikacin). For
patients with creatinine clearance < 60 mL/min, traditional dosing as described
below is recommended. With once-daily dosing, serum concentrations need not be
routinely checked until the second or third day of therapy, depending on the
stability of renal function and the anticipated duration of therapy. It is
unnecessary to check peak concentrations because they will be high. The goal is
to administer drug so that concentrations of less than 1 mcg/mL are present
between 18 and 24 hours after dosing. This provides a sufficient period of time
for washout of drug to occur before the next dose is given. Appropriate trough
levels can be accurately determined by measuring serum concentrations in
samples obtained 2 hours and 12 hours after dosing and then adjusting the dose
based on the actual clearance of drug or by measuring the concentration in a
sample obtained 8 hours after a dose. If the 8-hour concentra-tion is between
1.5 mcg/mL and 6 mcg/mL, the target trough can be achieved at 18 hours.
With traditional
dosing, adjustments must be made to prevent accumulation of drug and toxicity
in patients with renal insuffi-ciency. Either the dose of drug is kept constant
and the interval between doses is increased, or the interval is kept constant
and the dose is reduced. Nomograms and formulas have been constructed relating
serum creatinine levels to adjustments in treatment regi-mens. Because
aminoglycoside clearance is directly proportional to the creatinine clearance,
a method for determining the aminogly-coside dose is to estimate creatinine
clearance using the Cockcroft-Gault formula described. For a traditional twice-
or thrice-daily dosing regimen, peak serum concentrations should be determined
from a blood sample obtained 30–60 minutes after a dose, and trough
concentrations from a sample obtained just before the next dose. Doses of
gentamicin and tobramycin should be adjusted to maintain peak levels between 5
and 10 mcg/mL and trough levels < 2 mcg/mL (< 1 mcg/mL is optimal).
E. Adverse Effects
All
aminoglycosides are ototoxic and nephrotoxic. Ototoxicity and nephrotoxicity
are more likely to be encountered when therapy is continued for more than 5
days, at higher doses, in the elderly, and in the setting of renal
insufficiency. Concurrent use with loop diuretics (eg, furosemide, ethacrynic
acid) or other nephrotoxic antimicrobial agents (eg, vancomycin or
amphotericin) can poten-tiate nephrotoxicity and should be avoided if possible.
Ototoxicity can manifest either as auditory damage, resulting in tinnitus and
high-frequency hearing loss initially, or as vestibular damage, evi-dent by
vertigo, ataxia, and loss of balance. Nephrotoxicity results in rising serum
creatinine levels or reduced creatinine clearance, although the earliest indication
often is an increase in trough serum aminoglycoside concentrations. Neomycin,
kanamycin, and amikacin are the most ototoxic agents. Streptomycin and
gentamicin are the most vestibulotoxic. Neomycin, tobramycin, and gentamicin
are the most nephrotoxic.In very high doses, aminoglycosides can produce a curare-like
effect with neuromuscular blockade that results in respira-tory paralysis. This
paralysis is usually reversible by calcium gluconate (given promptly) or
neostigmine. Hypersensitivity occurs infrequently.
F. Clinical Uses
Aminoglycosides
are mostly used against gram-negative enteric bacteria, especially when the
isolate may be drug-resistant and when there is suspicion of sepsis. They are
almost always used in combination with a β-lactam antibiotic to extend coverage
to include potential gram-positive pathogens and to take advantage of the
synergism between these two classes of drugs. Penicillin-aminoglycoside
combinations also are used to achieve bactericidal activity in treatment of
enterococcal endocarditis and to shorten duration of therapy for viridans
streptococcal and some patients with staphylococcal endocarditis. Which
aminoglycoside and what dose should be used depend on the infection being
treated and the susceptibility of the isolate.
Related Topics