Chapter: Basic & Clinical Pharmacology : Antifungal Agents
Flucytosine
FLUCYTOSINE
Chemistry & Pharmacokinetics
Flucytosine (5-FC) was discovered in 1957 during a search for novel antineoplastic agents. Though devoid of anticancer proper-ties, it became apparent that it was a potent antifungal agent. Flucytosine is a water-soluble pyrimidine analog related to the chemotherapeutic agent 5-fluorouracil (5-FU). Its spectrum of action is much narrower than that of amphotericin B.
Flucytosine is currently available in North America only in an oral formulation. The dosage is 100–150 mg/kg/d in patients with normal renal function. It is well absorbed (> 90%), with serum concentrations peaking 1–2 hours after an oral dose. It is poorly protein-bound and penetrates well into all body fluid compart-ments, including the cerebrospinal fluid. It is eliminated by glom-erular filtration with a half-life of 3–4 hours and is removed by hemodialysis. Levels rise rapidly with renal impairment and can lead to toxicity. Toxicity is more likely to occur in AIDS patients and those with renal insufficiency. Peak serum concentrations should be measured periodically in patients with renal insuffi-ciency and maintained between 50 and 100 mcg/mL.
Mechanisms of Action & Resistanc
Flucytosine is taken up by fungal cells via the enzyme cytosine permease. It is converted intracellularly first to 5-FU and then to 5-fluorodeoxyuridine monophosphate (FdUMP) and fluorouridine triphosphate (FUTP), which inhibit DNA and RNA synthesis, respectively (Figure 48–1). Human cells are unable to convert the parent drug to its active metabolites, resulting in selective toxicity.
Synergy with amphotericin B has been demonstrated in vitro and in vivo. It may be related to enhanced penetration of the flu-cytosine through amphotericin-damaged fungal cell membranes. In vitro synergy with azole drugs has also been seen, although the mechanism is unclear.
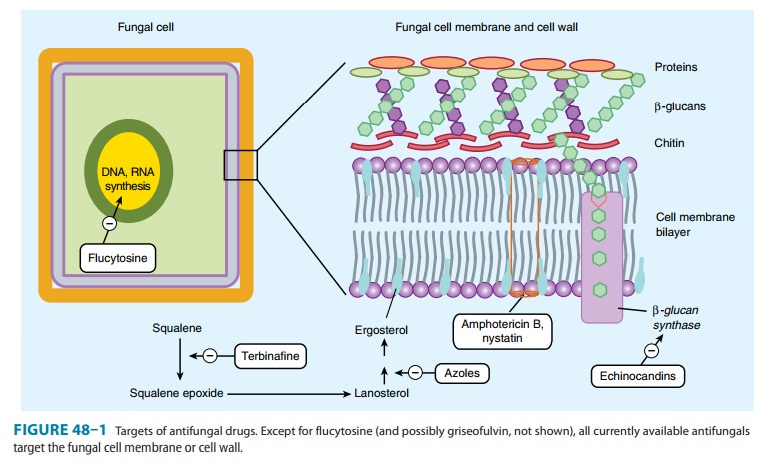
Resistance is thought to be mediated through altered metabo-lism of flucytosine, and, though uncommon in primary isolates, it develops rapidly in the course of flucytosine monotherapy.
Clinical Uses & Adverse Effects
The spectrum of activity of flucytosine is restricted to C neofor-mans, some Candida sp, and the dematiaceous molds that causechromoblastomycosis. Flucytosine is not used as a single agent because of its demonstrated synergy with other agents and to avoid the development of secondary resistance. Clinical use at present is confined to combination therapy, either with ampho-tericin B for cryptococcal meningitis or with itraconazole for chromoblastomycosis.
The adverse effects of flucytosine result from metabolism (possibly by intestinal flora) to the toxic antineoplastic compound fluorouracil. Bone marrow toxicity with anemia, leukopenia, and thrombocytopenia are the most common adverse effects, with derangement of liver enzymes occurring less frequently. A form of toxic enterocolitis can occur. There seems to be a narrow therapeutic window, with an increased risk of toxicity at higher drug levels and resistance developing rapidly at subtherapeutic concentrations. The use of drug concentration measurements may be helpful in reducing the incidence of toxic reactions, especially when flucytosine is com-bined with nephrotoxic agents such as amphotericin B.
Related Topics