Chapter: Basic & Clinical Pharmacology : Antifungal Agents
Azoles
AZOLES
Chemistry & Pharmacokinetics
Azoles are synthetic
compounds that can be classified as either imidazoles or triazoles according to
the number of nitrogen atoms in the five-membered azole ring, as indicated
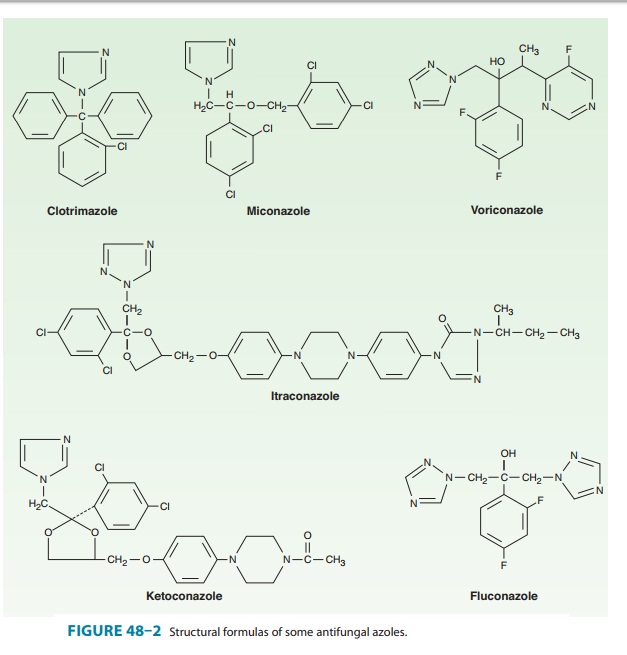
below. The imidazoles consist of ketoconazole, miconazole, and clotrimazole
(Figure 48–2). The latter two drugs are now used only in topical therapy. The
triazoles include itraconazole, fluconazole, voricon-azole, and posaconazole.
The pharmacology of
each of the azoles is unique and accounts for some of the variations in
clinical use. Table 48–2 summarizes the differences among five of the azoles.
Mechanisms of Action & Resistance
The antifungal
activity of azole drugs results from the reduction of ergosterol synthesis by
inhibition of fungal cytochrome P450 enzymes (Figure 48–1). The selective
toxicity of azole drugs results
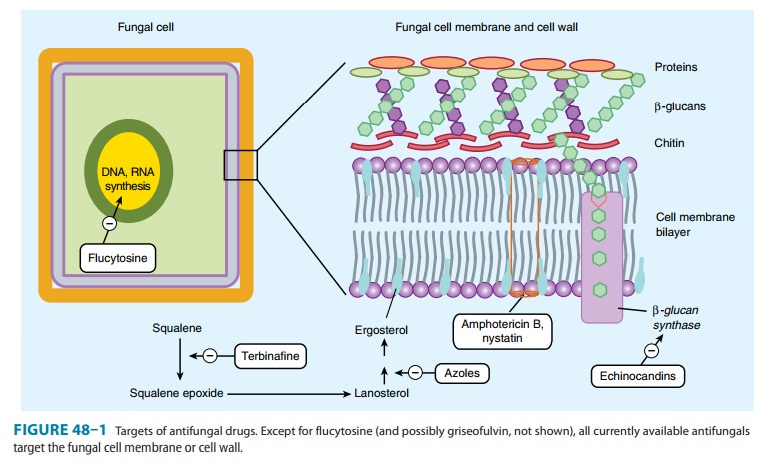
from their greater
affinity for fungal than for human cytochrome P450 enzymes. Imidazoles exhibit
a lesser degree of selectivity than the triazoles, accounting for their higher
incidence of drug interactions and adverse effects.
Resistance to azoles
occurs via multiple mechanisms. Once rare, increasing numbers of resistant
strains are being reported, suggest-ing that increasing use of these agents for
prophylaxis and therapy may be selecting for clinical drug resistance in
certain settings.
Clinical Uses, Adverse Effects, & Drug Interactions
The spectrum of action
of azole medications is broad, including many species of Candida, C neoformans, the endemic mycoses (blastomycosis,
coccidioidomycosis, histoplasmosis), the dermato-phytes, and, in the case of
itraconazole and voriconazole, even Aspergillus
infections. They are also useful in the treatment ofintrinsically
amphotericin-resistant organisms such as P
boydii.
As a group, the azoles
are relatively nontoxic. The most com-mon adverse reaction is relatively minor
gastrointestinal upset. All azoles have been reported to cause abnormalities in
liver enzymes and, very rarely, clinical hepatitis. Adverse effects specific to
indi-vidual agents are discussed below.
All
azole drugs are prone to drug interactions because they affect the mammalian
cytochrome P450 system of enzymes to some extent. The most significant
reactions are indicated below.
KETOCONAZOLE
Ketoconazole was the
first oral azole introduced into clinical use. It is distinguished from
triazoles by its greater propensity to inhibit mammalian cytochrome P450
enzymes; that is, it is less selective for fungal P450 than are the newer
azoles. As a result, systemic ketoconazole has fallen out of clinical use in
the USA and is not discussed in any detail here.
ITRACONAZOLE
Itraconazole
is available in oral and intravenous formulations and is used at a dosage of
100–400 mg/d. Drug absorption is increasedby food and by low gastric pH. Like
other lipid-soluble azoles, it interacts with hepatic microsomal enzymes,
though to a lesser degree than ketoconazole. An important drug interaction is
reduced bioavailability of itraconazole when taken with rifamycins (rifampin,
rifabutin, rifapentine). It does not affect mammalian steroid synthesis, and
its effects on the metabolism of other hepat-ically cleared medications are
much less than those of ketoconazole. While itraconazole displays potent
antifungal activity, effective-ness can be limited by reduced bioavailability.
Newer formula-tions, including an oral liquid and an intravenous preparation,
have utilized cyclodextran as a carrier molecule to enhance solubil-ity and
bioavailability. Like ketoconazole, itraconazole penetrates poorly into the
cerebrospinal fluid. Itraconazole is the azole of choice for treatment of
disease due to the dimorphic fungi Histoplasma, Blastomyces, and Sporothrix.
Itraconazole has activity against Aspergillus sp, but it has been replaced by
voriconazole as the azole of choice for aspergillosis. Itraconazole is used
extensively in the treatment of dermatophytoses and onychomycosis.
FLUCONAZOLE
Fluconazole
displays a high degree of water solubility and good cerebrospinal fluid
penetration. Unlike ketoconazole and itracon-azole, its oral bioavailability is
high. Drug interactions are also less common because fluconazole has the least
effect of all the azoles on hepatic microsomal enzymes. Because of fewer
hepatic enzyme interactions and better gastrointestinal tolerance, fluconazole
has the widest therapeutic index of the azoles, permitting more aggres-sive
dosing in a variety of fungal infections. The drug is available in oral and
intravenous formulations and is used at a dosage of 100–800 mg/d.
Fluconazole is the
azole of choice in the treatment and second-ary prophylaxis of cryptococcal
meningitis. Intravenous flucon-azole has been shown to be equivalent to
amphotericin B in treatment of candidemia in ICU patients with normal white
blood cell counts. Fluconazole is the agent most commonly used for the
treatment of mucocutaneous candidiasis. Activity against the dimorphic fungi is
limited to coccidioidal disease, and in particu-lar for meningitis, where high
doses of fluconazole often obviate the need for intrathecal amphotericin B.
Fluconazole displays no activity against Aspergillus
or other filamentous fungi.
Prophylactic use of
fluconazole has been demonstrated to reduce fungal disease in bone marrow
transplant recipients and AIDS patients, but the emergence of
fluconazole-resistant fungi has raised concerns about this indication.
VORICONAZOLE
Voriconazole is
available in intravenous and oral formulations. The recommended dosage is 400
mg/d. The drug is well absorbed orally, with a bioavailability exceeding 90%,
and it exhibits less protein binding than itraconazole. Metabolism is
predominantly hepatic. Voriconazole is a clinically relevant inhibitor of
mamma-lian CYP3A4, and dose reduction of a number of medications is required
when voriconazole is started. These include cyclosporine, tacrolimus, and
HMG-CoA reductase inhibitors. Observed toxicities include rash and elevated
hepatic enzymes. Visual dis-turbances are common, occurring in up to 30% of
patients receiv-ing intravenous voriconazole, and include blurring and changes
in color vision or brightness. These visual changes usually occur immediately
after a dose of voriconazole and resolve within 30 minutes. Photosensitivity
dermatitis is commonly observed in patients receiving chronic oral therapy.
Voriconazole
is similar to itraconazole in its spectrum of action, having excellent activity
against Candida sp (including
fluconazole-resistant species such as Candida
krusei) and the dimorphic fungi. Voriconazole is less toxic than
amphotericin B and is the treatment of choice for invasive aspergillosis.
POSACONAZOLE
Posaconazole is the
newest triazole to be licensed in the USA. It is available only in a liquid
oral formulation and is used at a dosage of 800 mg/d, divided into two or three
doses. Absorption is improved when taken with meals high in fat. Posaconazole
is rap-idly distributed to the tissues, resulting in high tissue levels but
relatively low blood levels. Visual changes have not been reported, but drug
interactions with increased levels of CYP3A4 substrates such as tacrolimus and
cyclosporine have been documented.
Posaconazole
is the broadest spectrum member of the azole family, with activity against most
species of Candida and Aspergillus. It is the only azole with
significant activity against the agents of mucormycosis. It is currently
licensed for salvage therapy in inva-sive aspergillosis, as well as prophylaxis
of fungal infections during induction chemotherapy for leukemia, and for
allogeneic bone marrow transplant patients with graft-versus-host disease.
Related Topics