Chapter: Biochemistry: Protein Purification and Characterization Techniques
Determining the Primary Structure of a Protein
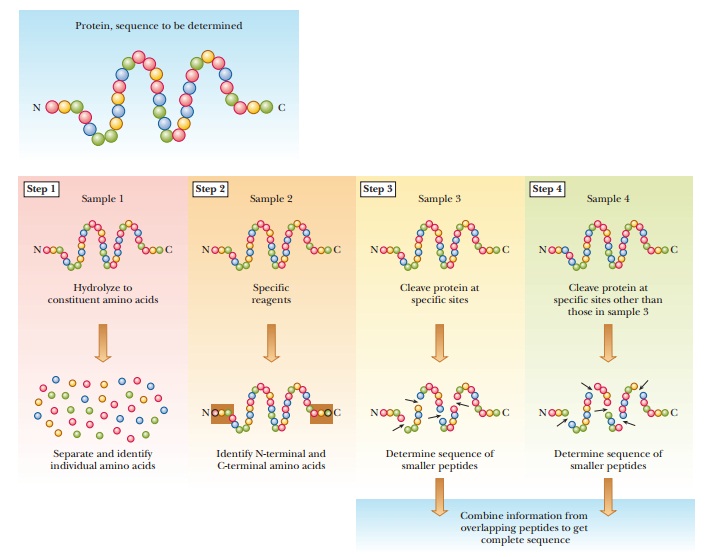
Determining the Primary Structure
of a Protein
Determining
the sequence of amino acids in a protein is a routine, but not trivial,
operation in classical biochemistry. Its several parts must be carried out
carefully to obtain accurate results (Figure 5.14).
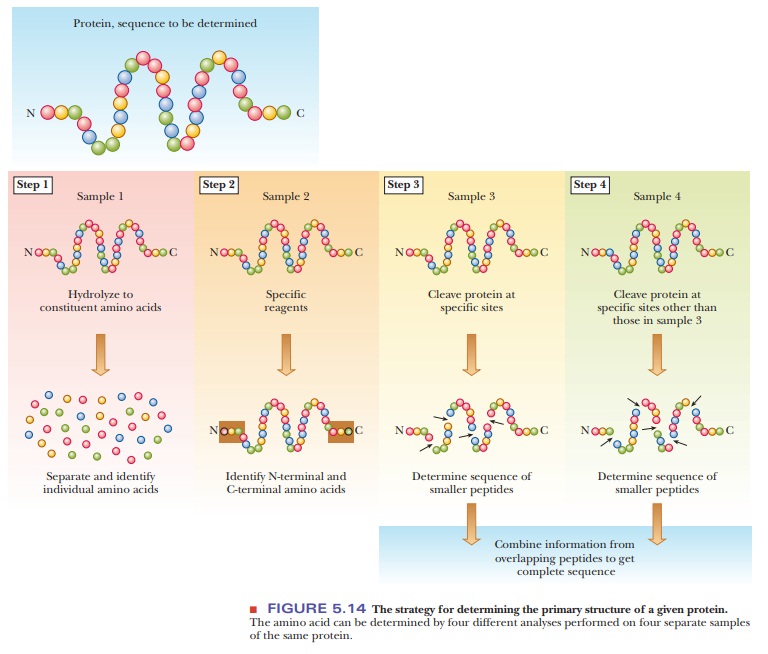
Step 1 in determining the primary structure of a protein is to establish which amino acids are present and in what proportions. Breaking a protein down to its component amino acids is relatively easy: heat a solution of the protein in acid, usually 6 M HCl, at 100°C to 110°C for 12 to 36 hours to hydrolyze the peptide bonds.
Separation and
identification of the products are somewhat more difficult and are best done by
an amino acid analyzer. This automated instrument gives both qualitative
information about the identities of the amino acids present and quantitative
information about the relative amounts of those amino acids. Not only does it
analyze amino acids, but it also allows informed decisions to be made about
which procedures to choose later in the sequenc-ing (see Steps 3 and 4 in
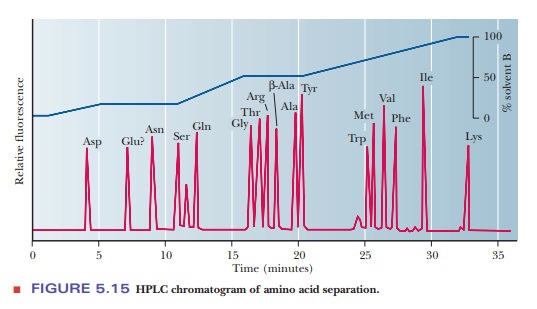
Figure 5.14). An amino acid analyzer separates the mixture of amino acids
either by ion-exchange chromatography or by high-performance liquid chromatography (HPLC), a chromatographic
techniquethat allows high-resolution separations of many amino acids in a short
time frame. Figure 5.15 shows a typical result of amino acid separation with
this technique.
In Step
2, the identities of the N-terminal and C-terminal amino acids in a protein
sequence are determined. This procedure is becoming less and less necessary as
the sequencing of individual peptides improves, but it can be used to check
whether a protein consists of one or two polypeptide chains.
In Steps
3 and 4, the protein is cleaved into smaller fragments, and the amino acid
sequence is determined. Automated instruments can perform a stepwise
modification starting from the N-terminal end, followed by cleav-age of each
amino acid in the sequence and the subsequent identification of each modified
amino acid as it is removed. This process is called the Edmandegradation.
Why are the proteins cleaved into small fragments for protein sequencing?
The
Edman degradation method becomes more difficult as the number of amino acids
increases. In most proteins, the chain is more than 100 residues long. For sequencing,
it is usually necessary to break a long polypeptide chain into fragments,
ranging from 20 to 50 residues for reasons that will be explained later.
Cleavage of the Protein into Peptides
Proteins
can be cleaved at specific sites by enzymes or by chemical reagents. The enzyme
trypsin cleaves peptide bonds
preferentially at amino acids that have positively charged R groups, such as
lysine and arginine.
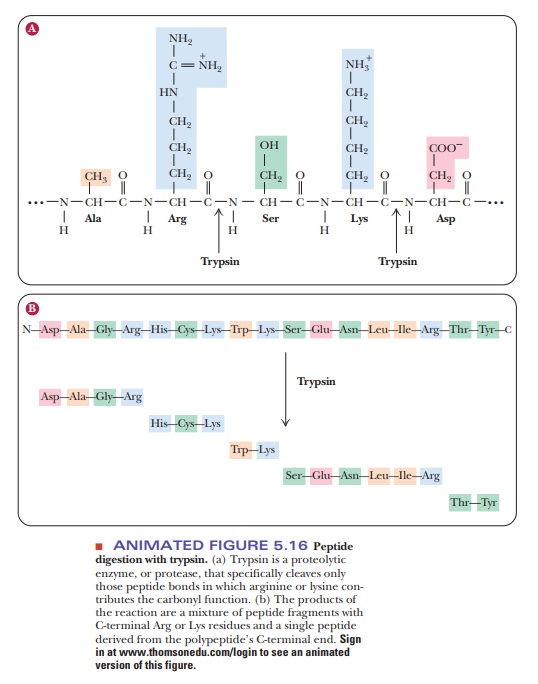
The cleavage takes place in
such a way that the amino acid with the charged side chain ends up at the
C-terminal end of one of the peptides produced by the reaction (Figure 5.16).
The C-terminal amino acid of the original protein can be any one of the 20
amino acids and is not necessarily one at which cleavage takes place. A peptide
can be automatically identified as the C-terminal end of the original chain if
its C-terminal amino acid is not a site of cleavage.
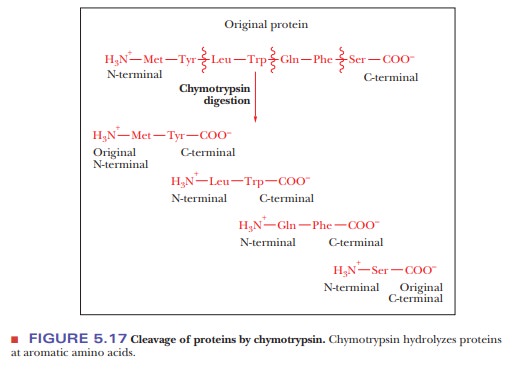
Another
enzyme, chymotrypsin, cleaves
peptide bonds preferentially at the aromatic amino acids: tyrosine, tryptophan,
and phenylalanine. The aromatic amino acid ends up at the C-terminal ends of
the peptides produced by the reaction (Figure 5.17).
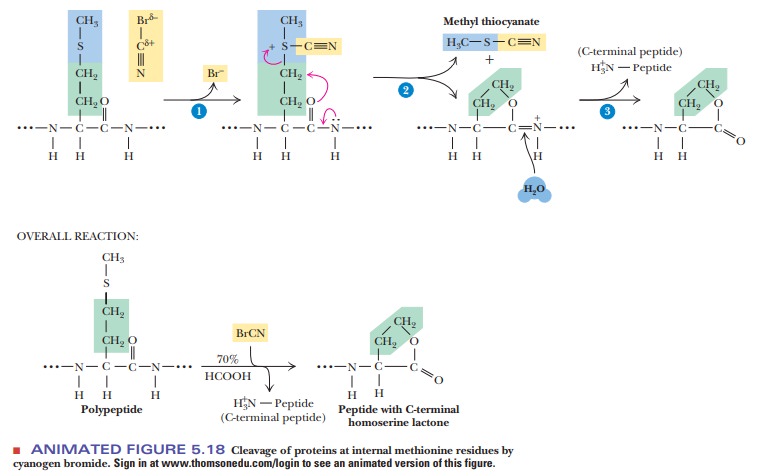
In the
case of the chemical reagent cyanogen
bromide (CNBr), the sites of cleavage are at internal methionine residues.
The sulfur of the methionine reacts with the carbon of the cyanogen bromide to
produce a homoserine lac-tone at the C-terminal end of the fragment (Figure
5.18).
The
cleavage of a protein by any of these reagents produces a mixture of peptides,
which are then separated by high-performance liquid chromatogra-phy. The use of
several such reagents on different samples of a protein to be sequenced
produces different mixtures. The sequences of a set of peptides produced by one
reagent overlap the sequences produced by another reagent (Figure 5.19). As a
result, the peptides can be arranged in the proper order after their own
sequences have been determined.

Sequencing of Peptides: The Edman Method
The actual sequencing of each peptide produced by specific cleavage of a protein is accomplished by repeated application of the Edman degradation.
The sequence of a peptide containing 10 to 40 residues can be determined by this method in about 30 minutes using as little as 10 picomoles of material, with the range being based on the amount of purified fragment and the complexity of the sequence.
For example, proline is more difficult to sequence than
serine because of its chemical reactivity. (The amino acid sequences of the
individual peptides in Figure 5.19 are determined by the Edman method after the
peptides are separated from one another.) The overlapping sequences of peptides
produced by different reagents provide the key to solving the puzzle. The
alignment of like sequences on different peptides makes deducing the overall
sequence possible. The Edman method has become so efficient that it is no
longer considered necessary to identify the N-terminal and C-terminal ends of a
protein by chemical or enzymatic methods. While interpreting results, however,
it is necessary to keep in mind that a protein may consist of more than one
polypeptide chain.

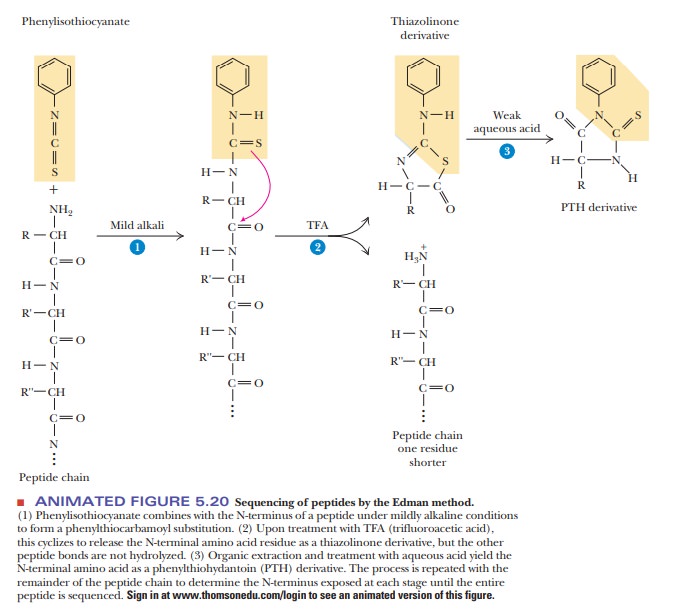
In the
sequencing of a peptide, the Edman reagent, phenyl
isothiocyanate, reacts with the peptide’s N-terminal residue. The modified
amino acid can be cleaved off, leaving
the rest of the peptide intact, and can be detected as the
phen-ylthiohydantoin derivative of the amino acid. The second amino acid of the
original peptide can then be treated in the same way, as can the third. With an
automated instrument called a sequencer
(Figure 5.20), the process is repeated until the whole peptide is sequenced.
Another
sequencing method uses the fact that the amino acid sequence of a protein
reflects the base sequence of the DNA in the gene that coded for that protein.
Using currently available methods, it is sometimes easier to obtain the
sequence of the DNA than that of the protein. Using the genetic code, one can
immediately determine the amino acid sequence of the protein. Convenient though
this method may be, it does not determine the positions of disulfide bonds or detect
amino acids, such as hydroxyproline, that are modi-fied after translation, nor
does it take into account the extensive processing that occurs with eukaryotic
genomes before the final protein is synthesized.


To
finish this section, let’s go back to why we needed to cut the protein into
pieces. Because the amino acid analyzer is giving us the sequence, it is easy
to think that we could analyze a 100-amino-acid protein in one step with the
ana-lyzer and get the sequence without having to digest the protein with
trypsin, chymotrypsin, or other chemicals. However, we must consider the logistical
reality of doing the Edman degradation. As shown in step 1 of Figure 5.20, we
react the peptide with the Edman reagent, phenylisothiocyanate (PITC). The
stoichiometry of this reaction is that one molecule of the peptide reacts with
one molecule of PITC. This yields one molecule of the PTH derivative in step 3
that is then analyzed. Unfortunately, it is very difficult to get an exact
stoichiometric match. For example, let’s say we are analyzing a peptide with
the sequence Asp-Leu-Tyr, etc. For simplicity, assume we add 100 molecules of
the peptide to 98 molecules of the PITC because we cannot measure the
quantities perfectly accurately. What happens then? In step 1, the PITC is
limiting, so we eventually end up with 98 PTH derivatives of aspartate, which
are analyzed correctly and we know the N-terminus is aspartate. In the second
round of the reaction, we add more PITC, but now there are two peptides; 98 of
them begin with leucine and 2 of them begin with aspartate. When we analyze the
PTH derivatives of round 2, we get two signals, one saying the derivative is
leucine and the other saying aspartate. In round 2, the small amount of PTH
derivative of aspartate does not interfere with our ability to recognize the
true second amino acid. However, with every round, this situation gets worse
and worse as more of the by-products show up. At some point, we get an analysis
of the PTH derivatives that cannot be identified. For this reason, we have to
start with smaller fragments so that we can analyze their sequences before the
signal degrades.
Summary
The amino acid sequence of a
protein can be determined using a multi-step process.
First, the protein is
hydrolyzed into its constituent amino acids and the composition determined.
The protein is also cleaved
into smaller fragments and these fragments are then sequenced by the Edman
degradation.
By using
overlapping fragments and the sequences determined, the sequence of the
original protein can be deduced.
Related Topics