Chapter: Basic & Clinical Pharmacology : Drugs of Abuse
Dependence: Tolerance & Withdrawal - Drugs of Abuse
DEPENDENCE: TOLERANCE &
WITHDRAWAL
With chronic exposure
to addictive drugs, the brain shows signs of adaptation. For example, if
morphine is used at short intervals, the dose has to be progressively increased
over the course of several days to maintain rewarding or analgesic effects.
This phenomenon is called tolerance. It may become a serious problem because of
increasing side effects—eg, respiratory depression—that do not show much
tolerance and may lead to fatalities associated with overdose.
Tolerance to opioids
may be due to a reduction of the concen-tration of a drug or a shorter duration
of action in a target system
Alternatively, it may involve changes of μ-opioid receptor function (pharmacodynamic
tolerance). In fact, many μ-opioid receptor agonists promote strong
receptor phosphorylation that triggers the recruitment of the adaptor pro-tein β-arrestin, causing G
proteins to uncouple from the receptor and to internalize within minutes .
Since this decreases signaling, it is tempting to explain tolerance by such a
mechanism. However, morphine, which strongly induces toler-ance, does not
recruit β-arrestins
and fails to promote receptor internalization. Conversely, other agonists that
drive receptor inter-nalization very efficiently induce only modest tolerance.
Based on these observations, it has been hypothesized that desensitization and
receptor internalization actually protect the cell from over-stimulation. In
this model, morphine, by failing to trigger receptor endocytosis,
disproportionally stimulates adaptive processes, whicheventually cause
tolerance. Although the molecular identity of these processes is still under
investigation, they may be similar to the ones involved in withdrawal .
Adaptive changes
become fully apparent once drug exposure is terminated. This state is called withdrawal and is observed to varying
degrees after chronic exposure to most drugs of abuse. Withdrawal from opioids
in humans is particularly strong (described below). Studies in rodents have
added significantly to our understanding of the neural and molecular mechanisms
that underlie dependence. For example, signs of dependence, as well as
analgesia and reward, are abolished in knockout mice lacking the μ-opioid receptor, but
not in mice lacking other opioid receptors(δ, κ). Although activation of the μ-opioid receptor
initially strongly inhibits adenylyl cyclase, this inhibition becomes weaker
after several days of repeated exposure.
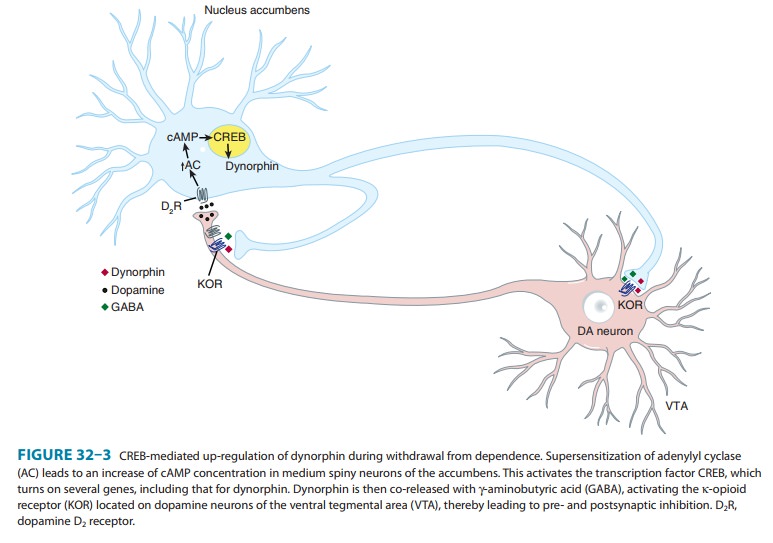
The reduction of the inhibition of adenylyl cyclase is due to a
counter-adaptation of the enzyme system during exposure to the drug, which
results in over-production of cAMP during subsequent withdrawal. Several
mechanisms exist for this adenylyl cyclase compensatory response, including
up-regulation of transcription of the enzyme. Increased cAMP concentrations in
turn strongly activate the transcription factor cyclic AMP response element
binding protein (CREB), leading to the regulation of downstream genes. Of the
few such genes identified to date, one of the most interesting is the gene for
the endogenous κ-opioid
ligand dynorphin. During withdrawal, neurons of the nucleus accumbens produce
high levels of dynor-phin, which is then co-released with GABA onto the
projection neurons of the VTA (Figure 32–3). These cells express κ-opioid receptors on
their synaptic terminals and on the dendrites. As a consequence, they are
inhibited and dopamine release is reduced. This mechanism exemplifies the
adaptive processes engaged during dependence and may underlie the intense
dysphoria typically observed during withdrawal.
Related Topics