Chapter: Pathology: Central Nervous System Pathology
Degenerative and Dementing Disorders
DEGENERATIVE AND DEMENTING DISORDERS
Parkinson disease (PD) is a progressive
neurodegenerative disease that involves genetic and environmental
factors. The SNCA gene (alpha-synuclein)
has been iden-tified as a risk factor, and gene mutations and multiplications
are associated with familial PD, but the majority of cases are sporadic. PD is
due to loss of dopaminergic neurons in the substantia nigra, leading to tremor,
rigidity, and akinesia.
•
Parkinson disease is the idiopathic form
•
Parkinson syndrome is secondary to known
injuries to the substantia nigra (e.g.,
infection, vascular condition, toxic insult).
Parkinson disease is common,
affecting 2% of the population. Clinical onset is typi-cally in decades 5–8.
Loss of dopaminergic neurons is still unexplained, though theories emphasize
oxidative stress. Pesticides and meperidine have been associated with increased
risk, while smoking and caffeine are protective.
On gross examination there is
pallor of the substantia nigra. Histology shows loss of pigmented
(dopaminergic) neurons in the substantia nigra. Residual neurons show Lewy
bodies, which are intracytoplasmic round eosinophilic inclusions that contain
α-synuclein. Electron microscopy shows filaments most likely of cytoskeletal
origin. There is also a secondary degeneration of dopaminergic axons in the
striatum.
Loss of the extrapyramidal
nigrostriatal pathway leads to inhibition of movement of proximal muscles and
disruption of fine regulation of distal muscles.
Involvement of the amygdala, cingulate gyrus and higher cortical regions causes
dementia and psychosis.
About 60% of patients
experience dementia 12 years after diagnosis; 50% also expe-rience depression
and psychosis. Those treated with medication (combination car-bidopa and
levodopa) and surgery (deep brain stimulation) will become refractory to
therapy.
A clinical diagnosis is
difficult to make early in disease because symptoms overlap with other
conditions. Early symptoms include hyposmia, constipation, and fatigue. Key
features are bradykinesia, rigidity, tremor and postural instability. Early in
the disease course, a response to levodopa can help confirm the diagnosis.
Imaging studies are not useful in most cases.
Huntington
disease (HD) is an autosomal dominant disorder. It is characterized pathologically by the
degeneration of GABAergic neurons of the caudate nucleus, and clinically by
involuntary movements, cognitive decline, and behavioral changes.
•
Affects those of northwestern European descent
•
Has an incidence in high-prevalence regions of 1/12,000–20,000
•
Gene (HTT), located on
chromosome 4, codes for a protein called huntingtin
• Mutations due to expansion of unstable cytosine-adenine-guanine
(CAG) repeats
• Shows features of anticipation and genomic imprinting
The pathophysiology is that
loss of caudate nucleus GABAergic neurons removes inhibitory influences on
extrapyramidal circuits, thus leading to chorea.
Clinical onset is typically
in decades 3–5. The chorea is characterized by sudden, unexpected, and
purposeless contractions of proximal muscles while awake. Psychi-atric symptoms
may predate motor symptoms. Disease progression leads to depen-dency and death.
Gross examination shows
atrophy of the caudate nucleus with secondary ventricular dilatation. Histology
shows loss of small neurons in the caudate nucleus followed by loss of the
larger neurons.
A definitive diagnosis can be
based on clinical symptoms with an affected parent. Otherwise, DNA
determination is the gold standard. Prenatal diagnosis and pre-implantation
diagnostics are available. Treatment is medical therapeutics for chorea
(dopamine receptor blocking or depleting agents).
Alzheimer disease (AD) causes 60% of all cases of dementia. It is the
most common cause of dementia in people age >65.
•
Incidence is 2% at age 65 and doubles every 5 years
•
Risk factors include aging and significant head trauma
Aluminum is an epiphenomenon, not a risk factor
•
Protective factors include high level of education and smoking
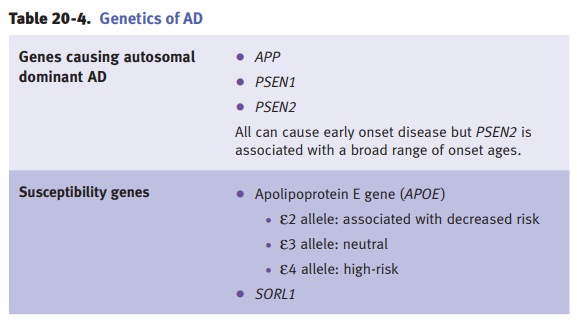
About 5–10% of AD cases are
hereditary, early onset, and transmitted as an autoso-mal dominant trait. There
are 3 genes that cause autosomal dominant AD:
•
APP (amyloid precursor protein)
•
Presenilin 1 and 2 (PSEN1 and 2)
Carriers of APP and PSEN1
mutations develop early- onset AD. Other AD suscepti-bility genes have been
identified. APOE is the largest effect locus for late-onset AD.
AD is characterized by
amyloid-β deposition, neurofibrillary angle formation, and neuronal
degeneration.
•
Abnormal proteins. Aβ amyloid is a 42-residue peptide derived from
a normal transmembrane protein, the amyloid precursor protein (APP). There is
also an abnormal tau (a microtubule-associated protein).
•
Neuritic plaques have a core of Aβ amyloid and are surrounded by
abnormal neurites.
•
Neurofibrillary tangles are intraneuronal aggregates of insoluble
cytoskeletal elements, mainly composed of abnormally phosphorylated tau forming
paired helical filaments.
•
Cerebral amyloid angiopathy is accumulation of Aβ amyloid within
the media of small and medium-size intracortical and leptomeningeal arteries;
it may occur by itself and cause intracerebral hemorrhage.
•
Additional changes include granulovacuolar degeneration and Hirano
bod-ies, which develop in the hippocampus and are less significant
diagnostically.
Affected areas are involved
in learning and memory. Lesions involve the neocortex, hippocampus, and several
subcortical nuclei including forebrain cholinergic nuclei (i.e., basal nucleus
of Meynert). The earliest and most severely affected are the hip-pocampus and
temporal lobe. Small numbers of neuritic plaques and neurofibrillary tangles
also form in intellectually normal aging persons.
Macroscopic changes include
atrophy of affected regions, producing brains that are smaller (atrophic), with
thinner gyri and wider sulci. Hippocampi and temporal lobes are markedly
atrophic.
Clinical manifestations have
insidious onset, typically beginning in decades 7–8. They include progressive
memory impairment, especially related to recent events; alterations in mood and
behavior; progressive disorientation; and aphasia (loss of language skills) and
apraxia (loss of learned motor skills). Within 5–10 years patients become mute
and bedridden.
No effective treatment is
available for AD but there is mild improvement with inhibi-tors of
acetylcholinesterase (e.g., tacrine).
Lewy body dementia is a
progressive brain disease associated with the formation of Lewy bodies in
neurons involving neocortex and subcortical nuclei. The etio-pathogenesis is
obscure, with no known risk factors; it is the second leading cause of
degenerative dementia in the elderly.
The histopathological
hallmark is the Lewy body. Neuron loss accompanies Lewy body formation. Sites
involved include the neocortex (especially the limbic system and cingulate
gyrus), and subcortical nuclei, including basal nucleus of Meynert, amygdala,
and substantia nigra.
The involvement of the
neocortex and substantia nigra is responsible for cogni-tive deterioration and
parkinsonism. Clinical manifestations include memory loss, parkinsonism, and
visual hallucinations. There is a possible treatment benefit from
cholinesterase inhibitors.
Amyotrophic lateral sclerosis (ALS) is the most common
adult-onset, progressive motor neuron disease.
The clinical diagnosis is
supported by a biopsy of muscles. The etiopathogenesis is obscure; 5–10% of
cases are hereditary, and a small number are caused by mutation of the gene
encoding zinc-copper superoxide dismutase on chromosome 21.
•
Loss of upper motor neurons produces hyperreflexia and
spasticity. In some cases,
involvement of cranial nerve nuclei also occurs.
•
Loss of lower motor neurons produces weakness, atrophy,
and fasciculations.
There is no cure for ALS.
Ultimately, involvement of respiratory muscles will lead to death.
Friedreich ataxia is an autosomal recessive
disorder which leads to degeneration of nerve tissue in the spinal
cord, especially those sensory neurons connected to the cerebellum affecting
muscle movement of the arms and legs. Onset is early child-hood.
Friedreich ataxia is caused
by the expansion of an unstable triplet nucleotide repeat (GAA repeats in the
first intron) in the frataxin gene on chromosome 9. The frataxin protein is
essential for mitochondrial function by helping in mitochondrial iron
regulation; in the absence of frataxin, mitochondrial iron builds up, leading
to free radical damage and mitochondrial dysfunction.
Clinical manifestations
include gait ataxia, dysarthria, hand clumsiness, loss of sense of position,
impaired vibratory sensation, and loss of tendon reflexes. There is an
increased incidence of heart disorders and diabetes. Patients become
wheelchair-bound by age 5.
Wilson disease
Acute intermittent porphyria is an autosomal dominant
defect in porphyrin metabo-lism with deficient uroporphyrinogen synthase. Both
porphobilinogen and ami-nolevulinic acid are increased. Urine is initially
colorless but on exposure to light turns dark red. Patients may develop
recurrent severe abdominal pain, psychosis, neuropathy, and dementia.
Vitamin B12 deficiency causes megaloblastic anemia,
demyelination of the spinal cord posterior columns and
lateral corticospinal tracts (subacute combined degen-eration of the spinal
tract). It also causes dementia and peripheral neuropathy.
Alcohol abuse causes generalized cortical
and cerebellar atrophy, as well as Wer-nicke-Korsakoff syndrome. The neurologic
disease is usually related to thiamine deficiency. There can be hemorrhages in
the mamillary bodies and the walls of the third and fourth ventricles. Neuronal
loss and gliosis may be prominent.
•
Wernicke encephalopathy has reversible confusion,
ataxia, and nystagmus.
•
Korsakoff
psychosis is more severe and has irreversible anterograde and ret-rograde
amnesia.
•
Central
pontine myelinolysis may cause death.
Related Topics