Chapter: Essential Clinical Immunology: Immunological Techniques
DNA Technology Assays
DNA Technology Assays
The emergence of molecular biology and DNA technology has ushered in a whole new series of methods for the detection of microbial agents, human genetics, and analysis of tissue samples, to name but a few of the uses of DNA technology today. The analysis of blood or serum samples also has far-reaching implications in our legal system. The following is a short sum-mary of these techniques and their impor-tance in clinical immunology.
DNA ANALYSIS
Known unique segments of nucleic acid sequences can be used as DNA probes to determine the presence of complemen-tary sequences of DNA in a sample from a given patient. The probe, which is a single strand of a given DNA, is presented to the target DNA, which is composed of thou-sands of nucleotides. The complementary strands from target and probe DNA will anneal to each other, a process known as DNA hybridization. The high affinity of the probe for a complementary segment in the target DNA is the most specific intermo-lecular interaction between biological mac-romolecules.
This technique may be used not only on fresh samples but also in tissues that have formalin-fixed and paraffin-embedded tissues. In this technique of in situ hybrid-ization, the probes can be applied directly to tissue sections on microscope slides. However, this technique works only after deparaffinization and proteolytic diges-tion are performed to expose intracellular nucleic acid targets. The probe is detected in the sample either by radiolabeling the probe or radiolabeling an antibody to the probe DNA.
Another approach is the use of restric-tion endonucleases (see Figure 2.6). These are enzymes that cleave DNA at sites specifi-cally related to the nucleotide sequences. Using enzymes with different specificities, a DNA fragment containing a particular
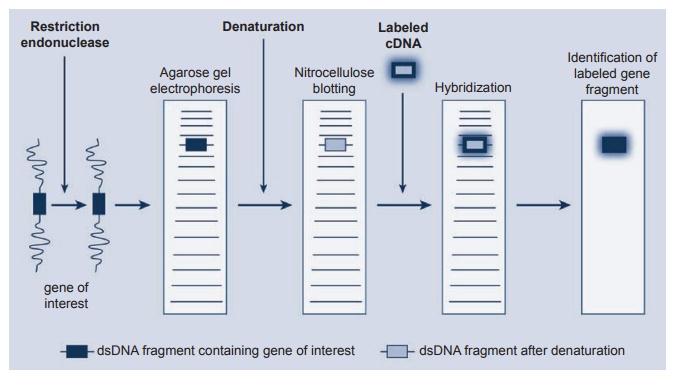
Figure 2.6 The use of restriction endonucleases to carry out gene mapping using the Southern blotting technique. DNA hybridization is the most specifi c intermolecular interaction between biological macromolecules.
gene can be cut out from the DNA mole-cule. In the Southern blot, these fragments of DNA are electophoresed on agarose gels with the smaller fragments migrating fur-ther than the larger ones. Alkaline dena-turation of these fragments uncoils the fragment so that a single stranded DNA will hybridize with the complementary DNA after transfer to a special nitrocellu-lose filter. Blotting the gel with the nitrocel-lulose filter “fixes” these DNA fragments after the electrophoresis. Finally, a radio-labeled probe containing DNA known to be complementary to the DNA of interest hybridizes to it, and the fragment can be identified by autoradiography of the filter. The Northern blotting technique is very similar to the DNA experiment but uses RNA instead of DNA in the system.
POLYMERASE CHAIN REACTION (PCR) ASSAY
Although the DNA analysis systems described earlier are still used in research and clinical laboratories, a major revolu
This method is particularly valuable since it can markedly amplify a small piece of DNA before cleavage with a restric-tion enzyme. Complementary oligonu-cleotide primers from either end of the target DNA are added to the denatured sample, along with a heat-resistant DNA polymerase. If the target sequence is pres-ent, the primers anneal to it and provide a starting point for the polymerase to begin the synthesis of second-strand DNA (see Figure 2.7). The newly synthesized double-stranded DNA is then denatured by heating and exposed again to the poly-merase enzyme at a lower temperature. In this way, newly synthesized molecules and original DNA can reassociate with the primer and act as templates for fur-ther rounds of DNA synthesis. After com-pleting about thirty cycles (usually two to three hours in an automated machine), the specific target sequence is amplified
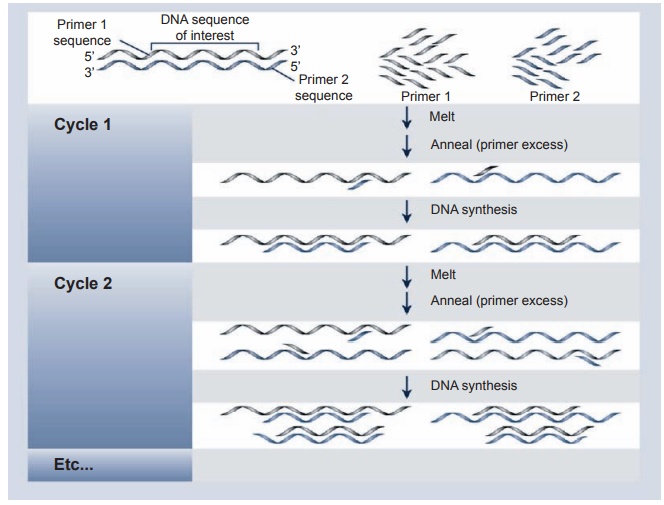
Figure 2.7 Schematic description of a PCR. To use this method, it is necessary to know the sequence of a short region of DNA on the 3′ and 5′ of the sequence. It is a very powerful tool for detecting DNA sequences in tissues, dried blood spots, and pathological specimens. The specimen must be handled carefully to avoid introduction of other DNA material, that is, the DNA of the handler of the specimen in question
more than 1-million-fold. This power-ful and sensitive technique can detect a specific DNA sequence from a single cell (e.g., lymphocyte, sperm), fixed pathologi-cal specimens, and dried blood spots. The main disadvantage is that contamination of the reaction mixture with traces of DNA from another source will lead to false positive results. Thus extreme care in handling specimens to be tested as well as in the test itself is obligatory.
MAJOR HISTOCOMPATIBILITY (MHC) ASSAYS
Using sera obtained first from multi-transfused patients and later from multipa-rous women (alloantigens), the early work of Dausset and others in the 1950s and
Using a somewhat crude agglutination assay of leucocytes, they noticed that these sera agglutinated leucocytes from unrelated donors and that the patterns of such co-agglutination dem-onstrated definitive antigens in human populations. After extensive research and workshop meetings, it became apparent that these antigens were present on all tis-sues of the body, but their high concen-tration on peripheral blood lymphocytes enabled investigators to perform immu-nogenetic studies more easily on these cells. While most of the original testing for the antigens on cells and for use in transplantation relied on serological tech-niques using purified antibodies to detect these antigens, the introduction of the PCR assay changed the rapidity with which these antigens could be identified, and both MHC class I and class II antigens are now routinely identified by the method.
Generally, this technique is available only in centers that specialize in organ and bone marrow transplantation. In addition to the transplantation, these techniques are used for familial genetic studies as well as for specific disease states. One example is the identification of HLA B27, which is highly associated with anklyosing spon-dylitis, and as the research continues one can expect that other genetic MHC mark-ers will be identified in particular disease states. The converse is also true. It is now known that certain MHC antigens confer resistance to a given disease, while others confer susceptibility. A word of caution is warranted. These markers only sug-gest a susceptibility to the disease. Most disease states appear to be polygenic, and factors such as the environment and past exposure to a given microbe also play a part in establishing the full disease pattern.
MICROARRAY ASSAYS
Over the past decade, this assay system has become popular in many branches of science, including immunology. In essence, these assays have become the standard tools for gene expression profiling as the mRNA levels of a large number of genes can be measured in a single assay. Of par-ticular interest, one can compare the lev-els of gene expressions across the human genome in one subset of patients in a given disease to another subset of patients with the same disease.
The method is relatively straightfor-ward. One takes the tissues or cells to be studied and prepares them as follows:
Tissue → mRNA → cDNA → Biotinylated cRNA
The biotinylated material is layered on glass slides on which DNA fragments of the human genome have been placed (12,000 probe sets/chips). The chips are then washed, stained with streptavidin-phycoerythrin, and scanned with a probe array scanner.
As an example, Figure 2.8 represents the staining patterns seen in six psoriatic patients before and six hours after treat-ment with a T-cell suppression agent. Note the decreased expression of red intensity in
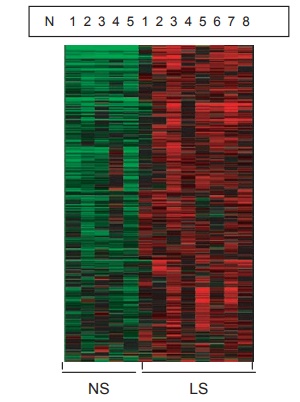
Figure 2.8 Photograph showing genomic analysis of lesional versus nonlesional skin in psoriatic skin. The heat map describes 1,119 genes that have signifi cantly higher expression (red area) by 1.2-fold (P < 0.05, after BH correction) in the lesional skin versus nonlesional skin (green area).
Related Topics