Chapter: Essential Clinical Immunology: Immunological Techniques
Immunological Assays
IMMUNOLOGICAL ASSAYS
Measurements of Immunoglobulins
The introduction of automated machines to measure immunoglobulins and other proteins has proceeded rapidly in recent decades. Most clinical immunology laboratories rely almost exclusively on these machines, and research labs are also intro-ducing these automated techniques at a rapid pace. Precise measurement of serum immunoglobulins is an essential corner-stone in this area and is important for repeated and serious infections secondary to immunosuppressive agents, immunode-ficiencies, in lymphoproliferate disorders, and for detection of autoantibodies.
The main principle behind this test is related to the formation of immune com-plexes between the antibody and a given antigen. If the concentration of antigen– antibody complex is low, then the immune complexes remain in suspension as fine particles, which can disperse a beam of light. As the complexes increase with concentration of antibody, the complexes will precipitate, and light scattering will decrease. This degree of dispersion can be measured on a nephelometer.
Using this method, a wide variety of proteins in serum, amniotic fluid, cerebro-spinal fluid, saliva, and gastrointestinal juices can be determined. The method includes a wide range of immune reac-tants, acute phase proteins, and tumor markers. Standard preparations are used and have been calibrated against interna-tional World Health Organization stan-dards. These tests primarily use polyclonal antibodies for each antigen since monoclo-nal antibodies do not form immune pre-cipitates because there are too few relevant epitopes.
Radioimmunoassay and Enzyme-Linked Immunosorbent Assays (ELISAs)
The use of these highly sensitive assays in human disease has virtually exploded in the past two decades. They can be used to detect the levels of a given antibody or hormone in human serum, and they are extremely sensitive methods of detecting low levels of autoantibodies.
In the radioimmunoassay, one can radiolabel a particular antigen or antibody using either 125I or 14C tagged to the antigen or antibody. Once the serum or purified antibody or antigen to be tested is placed in the well, a second radiolabeled antihuman IgG antibody is placed in the well. After appropriate binding and further washes, the degree of activity of the antibody to a given antigen can be determined in a γ counter (see Figure 2.1 top left).
The description of ELISAs; (Figure 2.1 top right) is similar to that described for the radioimmunoassay, but in place of the radioactively labeled antibody or anti-gen, various fluorochromes have been substituted in place of the radioactive label. In the presence of an appropriate substrate, the fluorochrome-labeled anti-body is activated to produce a given color, and the intensity of the color is read on a spectrophotometer using a 450-nm filter. By keeping the known antigen constant and diluting the serum to be tested, one can produce a curve of decreasing optical density readings, thereby indicating the amount of antibody in a given serum when compared with a standard control.
For detection of small amounts of a given antigen or antibody in a test sample, the “capture” assay is used (Figure 2.1 bot-tom). In this case, an unlabeled antibody to a given molecule is laid down on the plate to “capture” the small amount of antigen or antibody present in the test sample. The second antibody to this antigen or anti-body is labeled with the appropriate fluo-rochrome, and the rest of the tests proceed as in the direct assay described previously. While the radioimmunoassay remains the “gold” standard for many clinical labora-tories, more research and clinical laborato-ries are turning to the ELISA since it does not present the problem of radioactivity hazards or, perhaps more important, the removal of radioactive wastes (mainly a problem of disposal sites).
Immunoblots
This immunological technique has gained great favor with both basic immunolo-gists and clinical immunologists over the past decade. Its beauty is its simplicity and the fact that one can compare differ-ent proteins, toxins, and cellular products all at the same time and reach conclusions concerning their commonality or differ-ences or purity. The procedure is relatively simple. The proteins to be studied are run on a standard SDS gel, the percentage of which depends on the known or estimated size of the protein: larger proteins are run in 10 percent gels, while smaller proteins are run on 15 percent gels. The gel is then
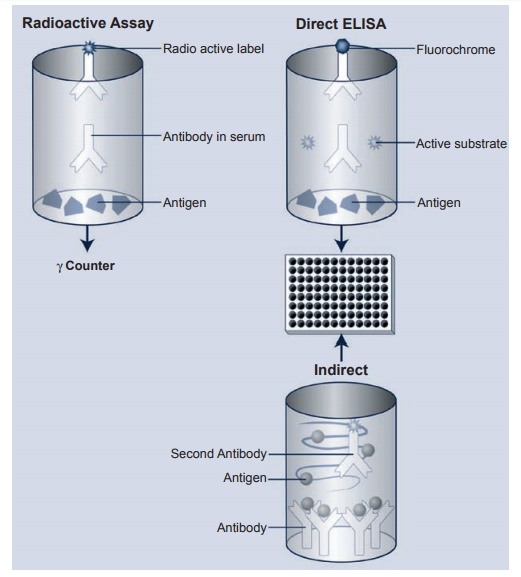
Figure 2.1 Schematic depiction of the radioimmunoassay and the direct and indirect ELISA. Both methods are extremely sensitive and will detect very small amounts of antibody in a given serum. One can label either antigen or antibody to determine amounts of antigen or antibody in a given serum or preparation. The indirect or sometimes called the “capture”. ELISA is also depicted.
removed and the proteins in the gel are transferred by another electrical charge to a cellulose membrane. The membrane is treated overnight with a blocking buffer, washed, and then layered over the mem-brane with the antibody designed to pick up the binding to the protein (S) in ques-tion. This incubation usually lasts one hour; following washes, the membrane is treated with a species-specific second anti-body tagged to an enzyme and developed with an enzyme substrate to form a colored band (see Figure 2.2).
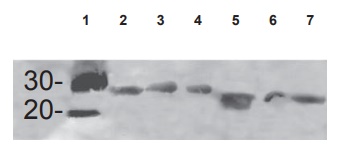
Figure 2.2 The photo is an example of an immunoblot in which the proteins in question are electrophoresed in a 15 percent SDS-gel, then transferred to nitrocellulose paper and incubated overnight in blocking buffer. This is followed by incubation with first antibody (1:1,000 dilution) followed by the species-specific second antibody tagged to alkaline phosphatase. The bands are developed using an alkaline phosphatase substrate. In this case, a single antipeptide antibody covering two sections of homology of the superantigens was used as the immunogen. Wells 2–7 contain three superantigens, each from the streptococcal and staphylococcal family of superantigens.
Complement Assays
Perhaps the most useful assays for comple-ment are the immunochemical assays of C3 and C4. As noted, which out-lines the alternative and classical modes of complement breakdown, a low C3 and C4 but normal factor B suggest that activa-tion of the classical pathways has occurred. Examples would be patients with systemic lupus erythematosus or vasculitis. In con-trast, if C3, C4, and factor B are all low, the alternative pathway is also activated
This would point to a gram-negative bacteremia. Normal C4 levels with low C3 and factor B levels suggest the alternative pathway alone. Elevation of all three components usually suggests acute or chronic infection. Acute rheumatic fever is such an example.
Assays for immune complexes are best directed toward an analysis of the immune complexes or their deposition in various human disease tissues. In most cases, the best approach is to receive freshly biop-sied nonfrozen material that is then snap frozen and sections cut and stained to test for the presence of appropriate antigen or antibody. In some cases, the antigen is still intact after formalin fixation and paraffin blocks are prepared. But in these specimens, one always runs the risk of destroying the appropriate antigen or antibody during the fixation process. Examples of the diseases studied in this manner are renal immune complex disease such as seen in SLE, acute poststreptococcal glomerulonephritis, or psoriasis.
Antibodies to common microbial anti-gens have been used to detect infection with different microbes for years. However, the presence of a single antibody serum specimen only tells you that the person was exposed to this microbe in the past. To diagnose an acute infection, one must have paired sera usually taken two weeks apart that demonstrate a significant rise in antibody titers in the second specimen compared to the first. One can also do these antibody tests in reverse. Normally, we are exposed to many microbial antigens during growth and development either as a result of exposure to a given microbe or after immunization with a given antigen (i.e., tetanus toxoid, pneumococcus polysac-charide, measles, or mumps viral antigen). Antibodies to the microbial products are usually found in normal individuals, but if they are not, one should suspect abnor-malities of antibody production like those seen in immunodeficiency states.
Related Topics