Chapter: Basic & Clinical Pharmacology : Vasodilators & the Treatment of Angina Pectoris
Calcium Channel-Blocking Drugs
CALCIUM CHANNEL-BLOCKING DRUGS
It
has been known since the late 1800s that transmembrane cal-cium influx is
necessary for the contraction of smooth and cardiac muscle. The discovery of a
calcium channel in cardiac muscle was followed by the finding of several different
types of calcium channels in different tissues (Table 12–4). The discovery of
these chan-nels made possible the measurement of the calcium current, ICa,
and subsequently, the development of clinically useful blocking
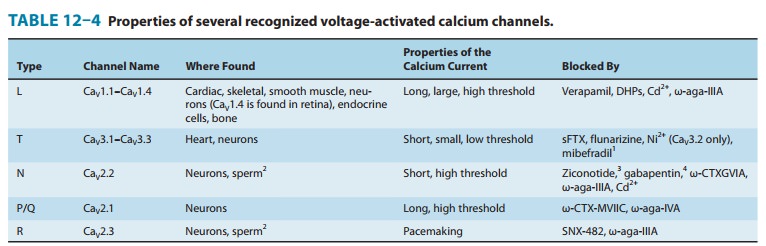
Although the blockers currently available for clinical use in cardiovascular
conditions are exclusively L-type calcium channel blockers, selective blockers
of other types of calcium channels are under intensive investigation. Certain
antiseizure drugs are thought to act, at least in part, through calcium channel
(espe-cially T-type) blockade in neurons .
Chemistry & Pharmacokinetics
Verapamil,
the first clinically useful member of this group, was the result of attempts to
synthesize more active analogs of papaverine, a vasodilator alkaloid found in
the opium poppy. Since then, doz-ens of agents of varying structure have been
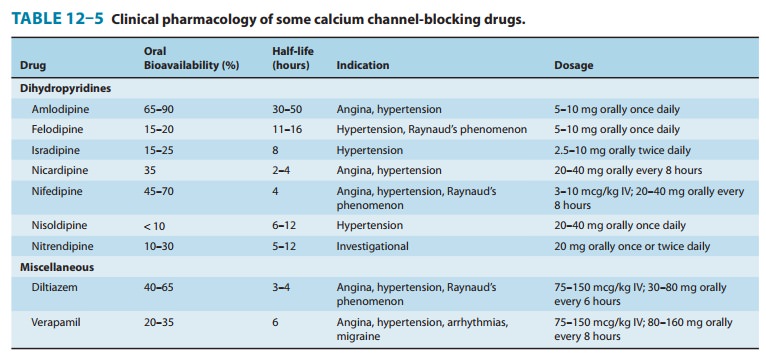
found to have the same fundamental pharmacologic action (Table 12–5). Three
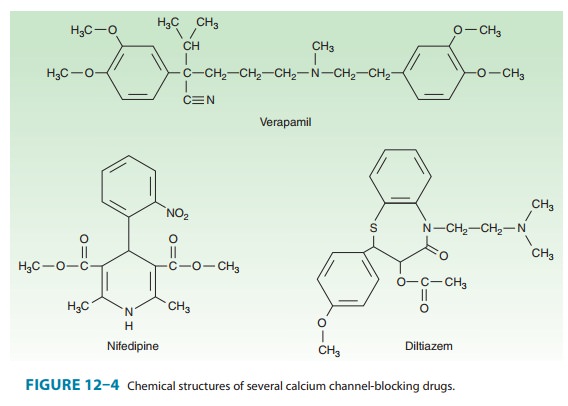
chemically dissimilar calcium channel blockers are shown in Figure 12–4.
Nifedipine is the prototype of the dihydropyridine family of calcium channel
blockers; dozens of molecules in this family have been investigated, and
several are currently approved in the USA for angina and other indications.
Nifedipine is the most extensively studied of this group, but the properties of
the other dihydropyridines can be assumed to be similar to it unless otherwise
noted.The calcium channel blockers are orally active agents and are
characterized by high first-pass effect, high plasma protein bind-ing, and
extensive metabolism. Verapamil and diltiazem are also used by the intravenous
route.
Pharmacodynamics
A. Mechanism of Action
The
voltage-gated L-type calcium channel is the dominant type in cardiac and smooth
muscle and is known to contain several drug receptors. It consists of α1 (the larger,
pore-forming sub-unit), α2, β, γ, and δ subunits. Four variant α1 subunits have been
recognized. Nifedipine and other dihydropyridines have been demonstrated to
bind to one site on the α1 subunit, whereas verapamil and diltiazem
appear to bind to closely related but not identical receptors in another region
of the same subunit. Binding of a drug to the verapamil or diltiazem receptors
allosterically affects dihydropyridine binding. These receptor regions are
ste-reoselective, since marked differences in both stereoisomer-bind-ing
affinity and pharmacologic potency are observed for enantiomers of verapamil,
diltiazem, and optically active nife-dipine congeners.
Blockade
of calcium channels by these drugs resembles that of sodium channel blockade by
local anesthetics. The drugs act from the inner side of the membrane and bind
more effectively to open channels and inactivated channels. Binding of the drug
reduces the frequency of opening in response to depolarization. The result is a
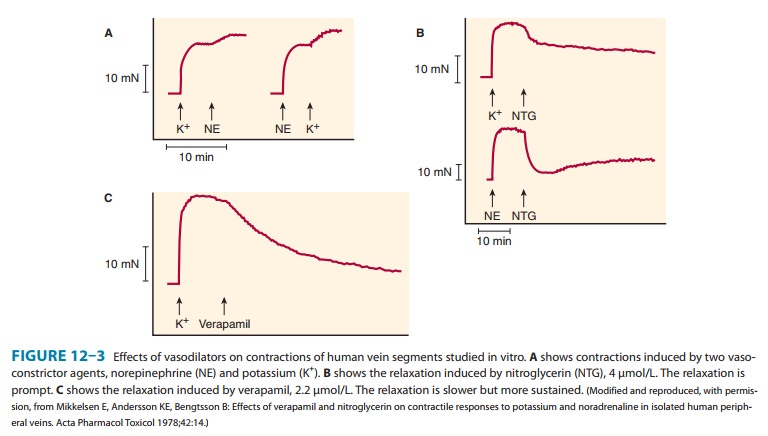
marked decrease in transmem-brane calcium current, which in smooth muscle
results in long-lasting relaxation (Figure 12–3) and in cardiac muscle results
in reduction in contractility throughout the heart and decreases in sinus node
pacemaker rate and atrioventricular node conduction velocity.∗ Although some neuronal cells harbor L-type
calcium channels, their sensitivity to these drugs is lower because the
chan-nels in these cells spend less time in the open and inactivated states.
Smooth
muscle responses to calcium influx through ligand-gated
calcium channels are also reduced by these drugs but not asmarkedly. The
block can be partially reversed by elevating the concentration of calcium,
although the levels of calcium required are not easily attainable in patients.
Block can also be partially reversed by the use of drugs that increase the
transmembrane flux of calcium, such as sympathomimetics.
Other
types of calcium channels are less sensitive to blockade by these calcium
channel blockers (Table 12–4). Therefore, tissues in which these other channel
types play a major role—neurons and most secretory glands—are much less
affected by these drugs than are cardiac and smooth muscle. Mibefradil is a selective T-type
calcium channel blocker that was introduced for antiar-rhythmic use but has
been withdrawn. Ion channels other than calcium channels are much less
sensitive to these drugs. Potassium channels in vascular smooth muscle are
inhibited by verapamil, thus limiting the vasodilation produced by this drug.
Sodium channels as well as calcium channels are blocked by bepridil, an obsolete antiarrhythmic drug.
B. Organ System Effects
1. Smooth muscle— Most types of smooth
muscle are depen-dent on transmembrane calcium influx for normal resting tone
and contractile responses. These cells are relaxed by the calcium channel
blockers (Figure 12–3). Vascular smooth muscle appears to be the most
sensitive, but similar relaxation can be shown for bronchiolar,
gastrointestinal, and uterine smooth muscle. In the vascular system, arterioles
appear to be more sensitive than veins; orthostatic hypotension is not a common
adverse effect. Blood pressure is reduced with all calcium channel blockers .
Women may be more sensitive than men to the hypotensive action of diltiazem.
The reduction in peripheral vascular resistance is one mechanism by which these
agents may benefit the patient with angina of effort. Reduction of coronary
artery spasm has been demonstrated in patients with variant anginaImportant
differences in vascular selectivity exist among the calcium channel blockers.
In general, the dihydropyridines have a greater ratio of vascular smooth muscle
effects relative to cardiac effects than do diltiazem and verapamil. The
relatively smaller effect of verapamil on vasodilation may be the result of
simultane-ous blockade of vascular smooth muscle potassium channels described
earlier. Furthermore, the dihydropyridines may differ in their potency in
different vascular beds. For example, nimodipine
is claimed to be particularly selective for cerebral blood vessels. Splice
variants in the structure of the α1 channel subunit appear to account for these
differences.
2. Cardiac muscle—Cardiac muscle is
highly dependent on cal-cium influx during each action potential for normal
function. Impulse generation in the sinoatrial node and conduction in the
atrioventricular node—so-called slow-response, or calcium-dependent, action
potentials—may be reduced or blocked by all of the calcium channel blockers.
Excitation-contraction coupling in all cardiac cells requires calcium influx,
so these drugs reduce cardiac contractility in a dose-dependent fashion. In
some cases, cardiac output may also decrease. This reduction in cardiac
mechanical function is another mechanism by which the calcium channel blockers
can reduce the oxygen requirement in patients with angina.
Important
differences between the available calcium channel blockers arise from the
details of their interactions with cardiac ion channels and, as noted above,
differences in their relative smooth muscle versus cardiac effects. Sodium
channel block is modest with verapamil, and still less marked with diltiazem.
It is negligible with nifedipine and other dihydropyridines. Verapamil and
diltiazem interact kinetically with the calcium channel receptor in a different
manner than the dihydropyridines; they block tachycardias in calcium-dependent
cells, eg, the atrioventricular node, more selec-tively than do the
dihydropyridines. On the other hand, the dihydropyridines appear to block
smooth muscle calcium channels at concentrations below those required for
significant cardiac effects; they are therefore less depres-sant on the heart
than verapamil or diltiazem.
3. Skeletal muscle—Skeletal muscle is not
depressed by thecalcium channel blockers because it uses intracellular pools of
calcium to support excitation-contraction coupling and does not require as much
transmembrane calcium influx.
4. Cerebral vasospasm and infarct
following subarach-noid hemorrhage—Nimodipine, a member of the dihydropyri-dine
group of calcium channel blockers, has a high affinity for cerebral blood
vessels and appears to reduce morbidity after a subarachnoid hemorrhage.
Nimodipine was approved for use in patients who have had a hemorrhagic stroke,
but it has recently been withdrawn. Nicardipine
has similar effects and is used by intravenous and intracerebral arterial
infusion to prevent cerebral vasospasm associated with stroke. Verapamil as
well, despite its lack of vasoselectivity, is used by the intra-arterial route
in stroke. Some evidence suggests that calcium channel blockers may also reduce
cerebral damage after thromboembolic stroke.
5. Other effects—Calcium channel
blockers minimally interferewith stimulus-secretion coupling in glands and nerve
endings because of differences between calcium channel type and sensitivity in
different tissues. Verapamil has been shown to inhibit insulin release in
humans, but the dosages required are greater than those used in management of
angina and other cardiovascular conditions.
A
significant body of evidence suggests that the calcium chan-nel blockers may
interfere with platelet aggregation in vitro and prevent or attenuate the
development of atheromatous lesions in animals. However, clinical studies have not
established their role in human blood clotting and atherosclerosis.
Verapamil
has been shown to block the P-glycoprotein respon-sible for the transport of
many foreign drugs out of cancer (and other) cells ; other calcium channel
blockers appear to have a similar effect. This action is not stereospecific.
Verapamil has been shown to partially reverse the resistance of cancer cells to
many chemotherapeutic drugs in vitro. Some clinical results sug-gest similar
effects in patients . Animal research suggests possible future roles of calcium
blockers in the treatment of osteoporosis, fertility disorders and male
contraception, immune modulation, and even schistosomiasis. Verapamil does not
appear to block transmembrane divalent metal ion transport-ers such as DMT1.
Toxicity
The
most important toxic effects reported for calcium channel blockers are direct
extensions of their therapeutic action. Excessive inhibition of calcium influx
can cause serious cardiac depression, including bradycardia, atrioventricular block,
cardiac arrest, and heart failure. These effects have been rare in clinical
use.
Retrospective
case-control studies reported that immediate-acting nifedipine increased the
risk of myocardial infarction in patients with hypertension. Slow-release and
long-acting dihydro-pyridine calcium channel blockers are usually well
tolerated. However, dihydropyridines, compared with angiotensin-converting
enzyme (ACE) inhibitors, have been reported to increase the risk of adverse
cardiac events in patients with hypertension with or without diabetes. These
results suggest that relatively short-acting calcium channel blockers such as
prompt-release nifedipine have the potential to enhance the risk of adverse
cardiac events and should be avoided. Patients receiving β-blocking drugs are
more sensitive to the cardiodepressant effects of calcium channel block-ers.
Minor toxicities (troublesome but not usually requiring dis-continuance of
therapy) include flushing, dizziness, nausea, constipation, and peripheral
edema. Constipation is particularly common with verapamil.
Mechanisms of Clinical Effects
Calcium
channel blockers decrease myocardial contractile force, which reduces
myocardial oxygen requirements. Calcium channel block in arterial smooth muscle
decreases arterial and intraven-tricular pressure. Some of these drugs (eg,
verapamil, diltiazem) also possess a nonspecific antiadrenergic effect, which
may con-tribute to peripheral vasodilation. As a result of all of these
effects, left ventricular wall stress declines, which reduces myocardial
oxy-gen requirements. Decreased heart rate with the use of verapamil or
diltiazem causes a further decrease in myocardial oxygen demand. Calcium
channel-blocking agents also relieve and pre-vent focal coronary artery spasm
in variant angina. Use of these agents has thus emerged as the most effective
prophylactic treat-ment for this form of angina pectoris.
Sinoatrial
and atrioventricular nodal tissues, which are mainly composed of
calcium-dependent, slow-response cells, are affected markedly by verapamil,
moderately by diltiazem, and much less by dihydropyridines. Thus, verapamil and
diltiazem decrease atrio-ventricular nodal conduction and are often effective
in the man-agement of supraventricular reentry tachycardia and in decreasing
ventricular responses in atrial fibrillation or flutter. Nifedipine does not
affect atrioventricular conduction. Nonspecific sympa-thetic antagonism is most
marked with diltiazem and much less with verapamil. Nifedipine does not appear
to have this effect. Significant reflex tachycardia in response to hypotension
occurs most frequently with nifedipine and less so with diltiazem and
verapamil. These differences in pharmacologic effects should be considered in
selecting calcium channel-blocking agents for the management of angina.
Clinical Uses of Calcium Channel-Blocking Drug
In
addition to angina, calcium channel blockers have well-documented efficacy in
hypertension and supraventricular
tachyarrhythmias . They also show moderate efficacy in a variety of other
conditions, including hypertrophic cardiomyopathy, migraine, and Raynaud’s
phenomenon. Nifedipine has some efficacy in preterm labor but is more toxic and
not as effective as atosiban, an
investigational oxytocin antagonist .
The
pharmacokinetic properties of these drugs are set forth in Table 12–5. The
choice of a particular calcium channel-blocking agent should be made with
knowledge of its specific potential adverse effects as well as its
pharmacologic properties. Nifedipine does not decrease atrioventricular
conduction and therefore can be used more safely than verapamil or diltiazem in
the presence of atrioventricular conduction abnormalities. A combination of
vera-pamil or diltiazem with β blockers may produce atrioventricular block
and depression of ventricular function. In the presence of overt heart failure,
all calcium channel blockers can cause further worsening of failure as a result
of their negative inotropic effect. Amlodipine,
however, does not increase mortality in patientswith heart failure due to
nonischemic left ventricular systolic dys-function and can be used safely in
these patients.
In
patients with relatively low blood pressure, dihydropyri-dines can cause
further deleterious lowering of pressure. Verapamil and diltiazem appear to
produce less hypotension and may be better tolerated in these circumstances. In
patients with a history of atrial tachycardia, flutter, and fibrillation,
verapamil and dilti-azem provide a distinct advantage because of their
antiarrhythmic effects. In the patient receiving digitalis, verapamil should be
used with caution, because it may increase digoxin blood levels through a
pharmacokinetic interaction. Although increases in digoxin blood level have
also been demonstrated with diltiazem and nifedipine, such interactions are
less consistent than with verapamil.
In
patients with unstable angina, immediate-release short-acting calcium channel
blockers can increase the risk of adverse cardiac events and therefore are
contraindicated (see Toxicity, above). However, in patients with non–Q-wave
myocardial infarc-tion, diltiazem can decrease the frequency of postinfarction
angina and may be used.
Related Topics