Chapter: Basic & Clinical Pharmacology : Antidepressant Agents
Basic Pharmacology of Antidepressants
BASIC PHARMACOLOGY OF
ANTIDEPRESSANTS
Chemistry & Subgroups
The currently
available antidepressants make up a remarkable variety of chemical types. These
differences and the differences in their molecular targets provide the basis
for distinguishing several subgroups.
A. Selective Serotonin Reuptake Inhibitors
The selective
serotonin reuptake inhibitors (SSRIs) represent a chemically diverse class of
agents that have as their primary action the inhibition of the serotonin
transporter (SERT) (Figure 30–3). Fluoxetine was introduced in the United
States in 1988 and quickly became one of the most commonly prescribed
medications in medical practice. The development of fluoxetine emerged out of
the search for chemicals that had high affinity for monoamine receptors but
lacked the affinity for histamine, ace-tylcholine, and α adrenoceptors that is seen with the tricyclic
antidepressants (TCAs). There are currently six available SSRIs, and they are
the most common antidepressants in clinical use. In addition to their use in
major depression, SSRIs have indications in GAD, PTSD, OCD, panic disorder,
PMDD, and bulimia. Fluoxetine,
sertraline, and citalopram exist
as isomers and areformulated in the racemic forms, whereas paroxetine and fluvoxamine are
not optically active. Escitalopram is
theSenantiomer of citalopram. As with
all antidepressants, SSRIs are highly lipophilic. The popularity of SSRIs stems
largely from their ease of use, safety in overdose, relative tolerability, cost
(all except escitalopram are generically available), and broad spec-trum of
uses.
B. Serotonin-Norepinephrine Reuptake Inhibitors
Two classes of
antidepressants act as combined serotonin and norepinephrine reuptake
inhibitors: selective serotonin-norepinephrine
reuptake inhibitors (SNRIs) and
TCAs.
Selective
serotonin-norepinephrine reuptake inhibitors - The SNRIs includevenlafaxine,its metabolitedesvenlafaxine,
and duloxetine. Another SNRI, milnacipran, has been approved for the
treatment of fibromyalgia in the USA but has been studied extensively as an
antidepressant. It has been available in Europe for several years. In addition
to their use in major depression, other applications of the SNRIs include the
treatment of pain disorders including neuropathies and fibromyalgia. SNRIs are
also used in the treatment of gen-eralized anxiety, stress urinary incontinence,
and vasomotor symptoms of menopause.
SNRIs are chemically
unrelated to each other. Venlafaxine was discovered in the process of
evaluating chemicals that inhibit binding of imipramine. Venlafaxine’s in vivo
effects are similar to those of imipramine but with a more favorable
adverse-effect profile. All SNRIs bind the serotonin (SERT) and norepinephrine
(NET) transporters, as do the TCAs. However, unlike the TCAs, the SNRIs do not
have much affin-ity for other receptors. Venlafaxine and desvenlafaxine are
bicyclic compounds, whereas duloxetine is a three-ring struc-ture unrelated to
the TCAs. Milnacipran contains a cyclopro-pane ring and is provided as a
racemic mixture.
Tricyclic
antidepressants—The TCAs were the
dominantclass of antidepressants until the introduction of SSRIs in the 1980s
and 1990s. Nine TCAs are available in the USA, and they all have an
iminodibenzyl (tricyclic) core (Figure 30–4). The chemical differences between
the TCAs are relatively subtle. For example, the prototype TCA imipramine and its metabolite, desipramine, differ by only a methyl
group in the propylamineside chain. However, this minor difference results in a
substantial change in their pharmacologic profiles. Imipramine is highly
anticholinergic and is a relatively strong serotonin as well as nor-epinephrine
reuptake inhibitor. In contrast, desipramine is much less anticholinergic and
is a more potent and somewhat more selective norepinephrine reuptake inhibitor
than is imipramine.
At the present time,
the TCAs are used primarily in depression that is unresponsive to more commonly
used antidepressants such as the SSRIs or SNRIs. Their loss of popularity stems
in large part from relatively poorer tolerability compared with newer agents,
to difficulty of use, and to lethality in overdose. Other uses for TCAs include
the treatment of pain conditions, enuresis, and insomnia.
C. 5-HT2 Antagonists
Two antidepressants
are thought to act primarily as antagonists at the 5-HT2 receptor: trazodone and nefazodone. Trazodone’s
structure includes a triazolo moiety that is thought to impart anti-depressant
effects. Its primary metabolite, m-chlorphenylpiperazine (m-cpp), is a potent
5-HT2 antagonist. Trazodone
was among the most commonly prescribed antidepressants until it was supplanted
by the SSRIs in the late 1980s. The most common use of trazodone in current
practice is as an unlabeled hypnotic, since it is highly sedating and not
associated with tolerance or dependence.
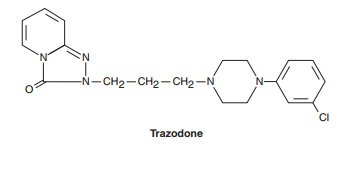
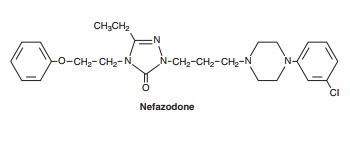
Nefazodone is chemically related to trazodone. Its primary metabolites, hydroxynefazodone and m-cpp are both inhibitors of the 5-HT2 receptor. Nefazodone received an FDA black box warning in 2001 implicating it in hepatotoxicity, including lethal cases of hepatic failure. Though still available generically, nefa-zodone is no longer commonly prescribed. The primary indica-tions for both nefazodone and trazodone are major depression, although both have also been used in the treatment of anxiety disorders.
D. Tetracyclic and Unicyclic Antidepressants
A number of
antidepressants do not fit neatly into the other classes. Among these are bupropion, mirtazapine, amoxapine, and maprotiline (Figure 30–5). Bupropion
has a unicyclic aminoketonestructure. Its unique structure results in a
different side-effect profile than most antidepressants (described below).
Bupropion somewhat resembles amphetamine in chemical structure and, like the
stimu-lant, has central nervous system (CNS) activating properties.Mirtazapine
was introduced in 1994 and, like bupropion, is one of the few antidepressants
not commonly associated with sexual side effects. It has a tetracyclic chemical
structure and belongs to the piperazino-azepine group of compounds.
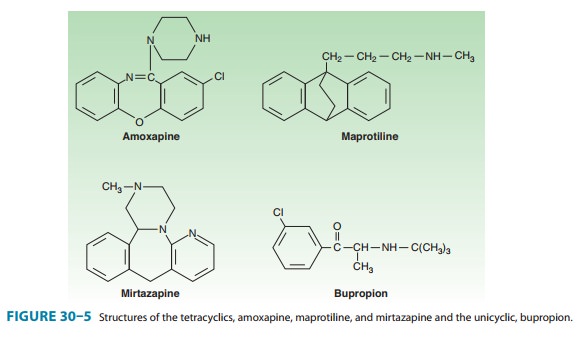
Mirtazapine,
amoxapine, and maprotiline have tetracyclic structures. Amoxapine is the N-methylated metabolite of loxa-pine, an
older antipsychotic drug. Amoxapine and maprotiline share structural
similarities and side effects comparable to the TCAs. As a result, these
tetracyclics are not commonly prescribed in current practice. Their primary use
is in MDD that is unre-sponsive to other agents.
E. Monoamine Oxidase Inhibitors
Arguably the first
modern class of antidepressants, monoamine oxidase inhibitors (MAOIs) were
introduced in the 1950s but are now rarely used in clinical practice because of
toxicity and poten-tially lethal food and drug interactions. Their primary use
now is in the treatment of depression unresponsive to other antidepres-sants.
However, MAOIs have also been used historically to treat anxiety states,
including social anxiety and panic disorder. In addi-tion, selegiline is used
for the treatment of Parkinson’s disease .
Current MAOIs include the hydrazine derivatives phenelzine and isocarboxazid and the non-hydrazines tranylcypromine,selegiline, and moclobemide (the latter is not available in theUSA). The hydrazines and tranylcypromine bind irreversibly and nonselectively with MAO-A and -B, whereas other MAOIs may have more selective or reversible properties. Some of the MAOIs such as tranylcypromine resemble amphetamine in chemical structure, whereas other MAOIs such as selegiline have amphet-amine-like metabolites. As a result, these MAOIs tend to have substantial CNS-stimulating effects.
Pharmacokinetics
The antidepressants
share several pharmacokinetic features (Table 30–1). Most have fairly rapid
oral absorption, achieve peak plasma levels within 2–3 hours, are tightly bound
to plasma pro-teins, undergo hepatic metabolism, and are renally cleared.
However, even within classes, the pharmacokinetics of individual
antidepressants varies considerably.
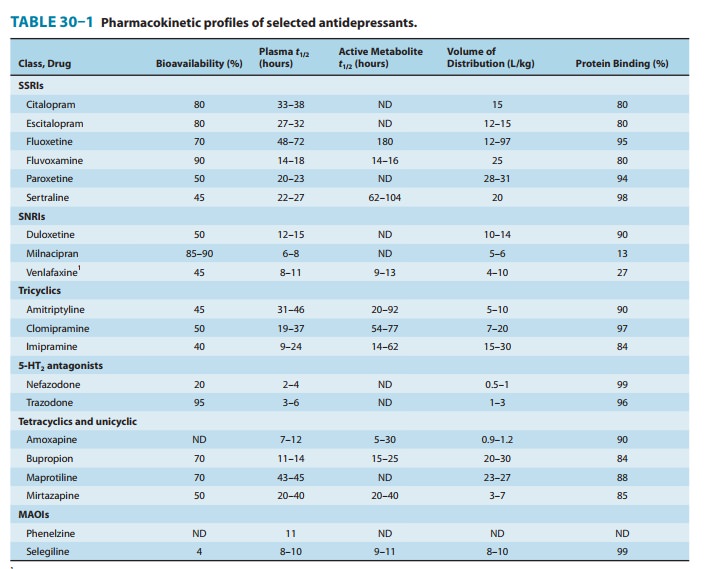
A. Selective Serotonin Reuptake Inhibitors
The prototype SSRI,
fluoxetine, differs from other SSRIs in some important respects (Table 30–1).
Fluoxetine is metabolized to an active product, norfluoxetine, which may have
plasma concentra-tions greater than those of fluoxetine. The elimination
half-life of norfluoxetine is about three times longer than fluoxetine and
contributes to the longest half-life of all the SSRIs. As a result,fluoxetine
has to be discontinued 4 weeks or longer before an MAOI can be administered to
mitigate the risk of serotonin syndrome.
Fluoxetine and
paroxetine are potent inhibitors of the CYP2D6 isoenzyme, and this contributes
to potential drug interactions (see Drug Interactions). In contrast,
fluvoxamine is an inhibitor of CYP3A4, whereas citalopram, escitalopram, and
sertraline have more modest CYP interactions.
B. Serotonin-Norepinephrine Reuptake Inhibitors
1. Selective serotonin-norepinephrine reuptake inhibitors—Venlafaxine is extensively metabolized in the livervia the CYP2D6 isoenzyme to O-desmethylvenlafaxine (desvenla-faxine). Both have similar half-lives of about 11 hours. Despite the relatively short half-lives, both drugs are available in formulations that allow once-daily dosing. Venlafaxine and desvenlafaxine have the lowest protein binding of all antidepressants (27–30%). Unlike most antidepressants, desvenlafaxine is conjugated and does not undergo extensive oxidative metabolism. At least 45% of desvenlafaxine is excreted unchanged in the urine compared with 4–8% of venlafaxine.
Duloxetine is well absorbed and has a half-life of about 12 hours but is dosed once daily. It is tightly bound to protein (97%) and undergoes extensive oxidative metabolism via CYP2D6 and CYP1A2. Hepatic impairment significantly alters duloxetine levels unlike desvenlafaxine.
2. Tricyclic antidepressants—The TCAs tend to be wellabsorbed and have long half-lives (Table 30–1). As a result, most are dosed once daily at night because of their sedating effects. TCAs undergo extensive metabolism via demethylation, aromatic hydroxylation, and glucuronide conjugation. Only about 5% of TCAs are excreted unchanged in the urine. The TCAs are substrates of the CYP2D6 system, and the serum levels of these agents tend to be substantially influenced by concurrent adminis-tration of drugs such as fluoxetine. In addition, genetic polymor-phism for CYP2D6 may result in low or extensive metabolism of the TCAs.
The secondary amine
TCAs, including desipramine and nor-triptyline, lack active metabolites and
have fairly linear kinetics. These TCAs have a wide therapeutic window, and
serum levels are reliable in predicting response and toxicity.
C. 5-HT2 Antagonists
Trazodone and
nefazodone are rapidly absorbed and undergo extensive hepatic metabolism. Both
drugs are extensively bound to protein and have limited bioavailability because
of extensive metabolism. Their short half-lives generally require split dosing
when used as antidepressants. However, trazodone is often prescribed as a
single dose at night as a hypnotic in lower doses than are used in the
treatment of depression. Both trazodone and nefazodone have active metabolites
that also exhibit 5-HT2 antagonism. Nefazodone is a potent inhibitor
of the CYP3A4 system and may interact with drugs metabolized by this enzyme
(see Drug Interactions).
D. Tetracyclic and Unicyclic Agents
Bupropion is rapidly
absorbed and has a mean protein binding of 85%. It undergoes extensive hepatic
metabolism and has a sub-stantial first-pass effect. It has three active
metabolites including hydroxybupropion; the latter is being developed as an
antidepres-sant. Bupropion has a biphasic elimination with the first phase
lasting about 1 hour and the second phase lasting 14 hours.
Amoxapine is also
rapidly absorbed with protein binding of about 85%. The half-life is variable,
and the drug is often given in divided doses. Amoxapine undergoes extensive
hepatic metabo-lism. One of the active metabolites, 7-hydroxyamoxapine, is a
potent D2 blocker and is associated with antipsychotic effects.
Maprotiline is similarly well absorbed orally and 88% bound to protein. It
undergoes extensive hepatic metabolism.
Mirtazapine is
demethylated followed by hydroxylation and glucuronide conjugation. Several CYP
isozymes are involved in the metabolism of mirtazapine, including 2D6, 3A4, and
1A2. The half-life of mirtazapine is 20–40 hours, and it is usually dosed once
in the evening because of its sedating effects.
E. Monoamine Oxidase Inhibitors
The different MAOIs
are metabolized via different pathways but tend to have extensive first-pass
effects that may substantially decrease bioavailability. Tranylcypromine is
ring hydroxylated and N-acetylated,
whereas acetylation appears to be a minor path-way for phenelzine. Selegiline
is N-demethylated and then
hydroxylated. The MAOIs are well absorbed from the gastrointestinal tract.
Because of the
prominent first-pass effects and their tendency to inhibit MAO in the gut
(resulting in tyramine pressor effects), alternative routes of administration
are being developed. For example, selegiline is available in both transdermal
and sublingualforms that bypass both gut and liver. These routes decrease the
risk of food interactions and provide substantially increased bioavail-ability.
Pharmacodynamics
As previously noted,
all currently available antidepressants enhance monoamine neurotransmission by
one of several mechanisms. The most common mechanism is inhibition of the
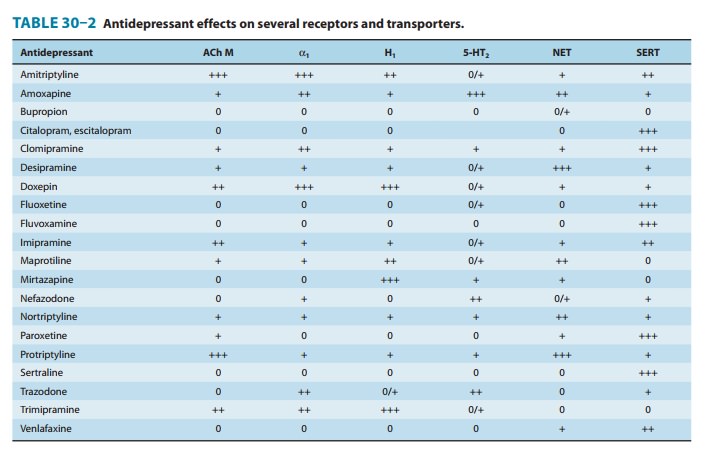
activity of SERT, NET, or both monoamine transporters (Table 30–2).
Antidepressants that inhibit SERT, NET, or both include the SSRIs and SNRIs (by
definition), and the TCAs. Another mechanism for increasing the availability of
monoam-ines is inhibition of their enzymatic degradation (the MAOIs).
Additional strategies for enhancing monoamine tone include binding presynaptic
autoreceptors (mirtazapine) or specific postsynaptic receptors (5-HT2 antagonists and
mirtazapine). Ultimately, the increased availability of monoamines for bind-ing
in the synaptic cleft results in a cascade of events that enhance the
transcription of some proteins and the inhibition of others. It is the net
production of these proteins, including BDNF, glucocorticoid receptors, β adrenoceptors, and
other proteins, that appears to determine the benefits as well as the toxicity
of a given agent.
A. Selective Serotonin Reuptake Inhibitors
The serotonin
transporter (SERT) is a glycoprotein with 12 trans-membrane regions embedded in
the axon terminal and cell body membranes of serotonergic neurons. When
extracellular serotonin binds to receptors on the transporter, conformational
changes occur in the transporter and serotonin, Na+, and Cl– are moved into the cell. Binding of
intracellular K+ then results in
return of the transporter to its original conformation and the release of
serotonin inside the cell. SSRIs allosterically inhibit the trans-porter by
binding the receptor at a site other than active binding site for serotonin. At
therapeutic doses, about 80% of the activity of the transporter is inhibited.
Functional polymorphisms exist for SERT that determine the activity of the
transporter.
SSRIs have modest
effects on other neurotransmitters. Unlike TCAs and SNRIs, there is little
evidence that SSRIs have promi-nent effects on β adrenoceptors or the norepinephrine
transporter, NET. Binding to the serotonin transporter is associated with tonic
inhibition of the dopamine system, although there is substantial
interindividual variability in this effect. The SSRIs do not bind aggressively
to histamine, muscarinic, or other receptors.
B. Drugs That Block Both Serotonin and Norepinephrine Transporters
A large number of
antidepressants have mixed inhibitory effects on both serotonin and
norepinephrine transporters. The newer agents in this class (venlafaxine and
duloxetine) are denoted by the acronym SNRIs, whereas the agents in the older
group are termed TCAs.
1. Serotonin-norepinephrine
reuptake inhibitors—SNRIsbind both the
serotonin and the norepinephrine transporters. The NET is structurally very
similar to the 5-HT transporter. Like the serotonin transporter, it is a
12-transmembrane domain complex that allosterically binds norepinephrine. The
NET also has a mod-erate affinity for dopamine.
Venlafaxine is a weak
inhibitor of NET, whereas desvenlafaxine, duloxetine, and milnacipran are more
balanced inhibitors of both SERT and NET. Nonetheless, the affinity of most
SNRIs tends tobe much greater for SERT than for NET. The SNRIs differ from the
TCAs in that they lack the potent antihistamine, α-adrenergic blocking, and anticholinergic
effects of the TCAs. As a result, the SNRIs tend to be favored over the TCAs in
the treatment of MDD and pain syndromes because of their better tolerability.
2. Tricyclic antidepressants—The TCAs resemble the SNRIsin function, and
their antidepressant activity is thought to relate primarily to their
inhibition of 5-HT and norepinephrine reuptake. Within the TCAs, there is
considerable variability in affinity for SERT versus NET. For example,
clomipramine has relatively very little affinity for NET but potently binds
SERT. This selectivity for the serotonin transporter contributes to
clomipramine’s known benefits in the treatment of OCD. On the other hand, the
second-ary amine TCAs, desipramine and nortriptyline, are relatively more
selective for NET. Although the tertiary amine TCA imipramine has more
serotonin effects initially, its metabolite, desipramine, then balances this
effect with more NET inhibition.Common adverse effects of the TCAs, including
dry mouth and constipation, are attributable to the potent antimuscarinic
effects of many of these drugs. The TCAs also tend to be potent antagonists of
the histamine H1 receptor. TCAs such
as doxepin are sometimes prescribed as hypnotics and used in treatments for
pruritus because of their antihistamine properties. The blockade of α adrenoceptors can
result in substantial orthostatic hypoten-sion, particularly in older patients.
C. 5-HT2 Antagonists
The principle action
of both nefazodone and trazodone appears to be blockade of the 5-HT2A
receptor. Inhibition of this receptor in both animal and human studies is
associated with substantial antianxiety, antipsychotic, and antidepressant
effects. Conversely, agonists of the 5-HT2A receptor, eg, lysergic
acid (LSD) and mes-caline, are often hallucinogenic and anxiogenic. The 5-HT2A
receptor is a G protein-coupled receptor and is distributed throughout the
neocortex.
Nefazodone is a weak
inhibitor of both SERT and NET but is a potent antagonist of the postsynaptic
5-HT2A receptor, as are its
metabolites. Trazodone is also a weak but selective inhibitor of SERT with
little effect on NET. Its primary metabolite, m-cpp, is a potent 5-HT2 antagonist, and much
of trazodone’s benefits as an antidepressant might be attributed to this
effect. Trazodone also has weak-to-moderate presynaptic α-adrenergic–blocking
proper-ties and is a modest antagonist of the H1 receptor.
D. Tetracyclic and Unicyclic
Antidepressants
The actions of
bupropion remain poorly understood. Bupropion and its major metabolite
hydroxybupropion are modest-to-mod-erate inhibitors of norepinephrine and
dopamine reuptake in animal studies. However, these effects seem less than are
typically associated with antidepressant benefit. A more significant effect of
bupropion is presynaptic release of catecholamines. In animal studies,
bupropion appears to substantially increase the presynap-tic availability of
norepinephrine, and dopamine to a lesser extent. Bupropion has virtually no
direct effects on the serotonin system.
Mirtazapine has a
complex pharmacology. It is an antagonist of the presynaptic α2 autoreceptor and
enhances the release of both norepinephrine and 5-HT. In addition, mirtazapine
is an antago-nist of 5-HT2 and 5-HT3 receptors. Finally, mirtazapine is a potent H1 antagonist, which is
associated with the drug’s sedative effects.
The actions of
amoxapine and maprotiline resemble those of TCAs such as desipramine. Both are
potent NET inhibitors and less potent SERT inhibitors. In addition, both
possess anticholin-ergic properties. Unlike the TCAs or other antidepressants,
amox-apine is a moderate inhibitor of the postsynaptic D2 receptor. As such,
amoxapine possesses some antipsychotic properties.
E. Monoamine Oxidase Inhibitors
MAOIs act by mitigating the actions of monoamine oxidase in the neuron and increasing monoamine content. There are two forms of monoamine oxidase. MAO-A is present in both dopamine and norepinephrine neurons and is found primarily in the brain, gut, placenta, and liver; its primary substrates are norepinephrine,epinephrine, and serotonin. MAO-B is found primarily in sero-tonergic and histaminergic neurons and is distributed in the brain, liver, and platelets. MAO-B acts primarily on tyramine, phenyl-ethylamine, and benzylamine. Both MAO-A and -B metabolize tryptamine and dopamine.
MAOIs are classified
by their specificity for MAO-A or -B and whether their effects are reversible
or irreversible. Phenelzine and tranylcypromine are examples of irreversible,
nonselective MAOIs. Moclobemide is a reversible and selective inhibitor of
MAO-A but is not available in the USA. Moclobemide can be displaced from MAO-A
by tyramine, and this mitigates the risk of food interac-tions. In contrast,
selegiline is an irreversible MAO-B–specific agent at low doses. Selegiline is
useful in the treatment of Parkinson’s disease at these low doses, but at
higher doses it becomes a nonselective MAOI similar to other agents.
Related Topics