Chapter: Essential Clinical Immunology: Basic Components of the Immune System
Antibody Production
ANTIBODY PRODUCTION
To achieve antibody production, at least four types of cells are required: APC, B cells, and two types of regulating cells.
B Cells
Antibodies are produced by naïve B cells and are called plasma cells. These cells express immunoglobulins on their sur-face. In the early stages, B cells first show intracellular µ-chains and then surface IgM. Through the process described ear-lier, these cells can later express IgG, IgA, or IgE, a phenomenon known as isotype switching. The final type of surface immu-noglobulin determines the class of anti-body secreted.
Isotype switching is mediated through two important protein interactions: CD40 on the B cell interacts with CD40L on acti-vated T cells (IL-4 induced) to stimulate B
Deficiencies in either molecule lead to severe immunodeficiency states with only IgM produced but no IgG or IgA antibodies. This syndrome is called the hyper-IgM syndrome, and in this case of CD40L deficiency, it is an X-linked immu-nodeficiency.
As mentioned before, each B cell is committed to the production of antibody expressing a unique VH–VL combination, and the surface and excreted immuno-globulin are the same. These observations form the basis of Burnet’s clonal selection theory in that each B cell expresses a sur-face immunoglobulin that acts as its anti-gen-receptor site. Contact with the antigen and helper T-cell factors commit each B cell to divide and differentiate to pro-duce more of the same VH–VL antibody. A number of these B cells become memory cells so that a greater number of antigen-specific B cells are available on a second-ary contact with the same antigen. This phenomenon is known as clonal expan-sion and helps to account for the greater secondary response.
Perhaps more important is that the sec-ondary response of antibodies has a higher affinity binding for these antigens. These latter antibodies will bind to antigen even when complexed to antibody and help clear the antigen more effectively from the circulation. It is important to remember, however, that B cells alone do not respond to antigen directly, even in the presence of APC cells. They must have a second signal, normally provided by the T cells. This point was elegantly shown in a series of transfer experiments using irradiated recipient ani-mals. As seen in Figure 1.8, antigen alone or antigen + B cells produced no antibody production in these animals. Similarly, T cells alone were ineffective. However,
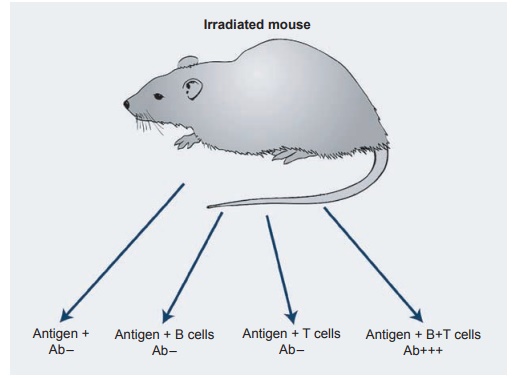
Figure 1.8 Adoptive cell transfer experiments in irradiated animals shows both T and B cells important for antibody production; Ab, antibody.
T Cells
One must first emphasize that helper T cells can only respond to antigen presented by macrophage MHC class II antigens as a complex on APC cells. In turn, they recog-nize the same combination of antigen and class II MHC antigens on the correspond-ing B cells. It is only then that the helper T cell secretes its cytokines to activate the reaction. As seen in Figure 1.8, T cells rec-ognize antigen in the context of their own MHC configuration. They will not cooper-ate with B cells and macrophages express-ing antigens of a different genetic back-ground.
When helper T cells meet an antigen for the first time, only a limited number of cells are activated to provide help for the B cells. However, when the animal is re-exposed, there is a marked increase of specific helper T cells. These cells constitute an expanded clone and the immune response is quicker and more vigorous.
This activation is also aided by two other mechanisms. First, memory cells have increased numbers of adhesion mol-ecules (LFA-1, CD2, LFA-3, and ICAM-1) on their surface as well as a higher popu-lation of affinity receptors. Thus, memory cells produce high concentrations of IL-2 to recruit more helper cells of both types TH1 and TH2. The recognition of antigen involves several receptors on the surface of the T cells. In contrast, B cells recognize antigen by surface-bound immunoglobulin and recognize the same epitopes somewhat differently. T cells only recognize haptens (small molecules) when the haptens are coupled to a carrier protein while antibody can recognize free haptens easily.
Differentiation of T Cells
T cells have characteristic cell-surface glycoproteins that serve as markers of“differentiation” of these cells. These markers are recognized by specific mono-clonal antibodies and divide them into two particular subsets.
TH1 cells secrete TNF and INF-α and mediate cellular immunity. Conversely, TH2 cells secrete IL-4, IL-5, IL-10, and IL-13 and are needed for stimulating antibody production by B cells. T cells secreting both cytokine profiles are designated THO.
What influences a naïve T cell to select which cytokine profile to secrete is not known. However, experiments in which cells are exposed to IL-1 and IL-6 promote TH2 cells while IL-12 and IFN-α stimulate production of TH1 T cells.
In humans, a TH1 cytokine profile is primarily directed toward protection against intracellular pathogens while a TH2 profile interacts with diseases charac-terized by overproduction of antibodies including IgE.
An elegant example of these different pathways of the TH1 and TH2 response is seen in the disease leprosy. Patients that develop a TH1 response develop only limited disease (tuberculoid leprosy). In contrast, those patients mounting a TH2 response develop debilitating and spread-ing lepromatous leprosy since the antibody response will not protect against an intra-cellular pathogen
Cellular Immunity
Cell-mediated responses are implemented by T lymphocytes. The major functions of T cells can be divided into two catego-ries: the first (cytotoxicity) is to lyse cells expressing specific antigens; the second (delayed hypersensitivity) is to release cytokines, thereby triggering an inflam-matory response. These two types of cells are used to combat intracellular pathogenssuch as viruses, certain bacteria, and para-sites inaccessible to antibodies.
Cytotoxic T cells lyse cells infected with viruses. This cytotoxicity is virus specific, and only cells expressing those proteins on the surface of the infected cell are killed. As stated before, this destruction occurs only in the presence of the same MHC class I molecules. This combination directly acti-vates CD8+ cells and is a potent killer of virally infected cells. The induction of the cytotoxic T cell requires precursor cells and IL-2 from helper cells and is subject to regulation by other T cells.
Cytotoxic T cells also play a role in graft rejection. This was shown years ago in a mixed lymphocyte reaction in which the lymphocytes from two genetically differ-ent individuals were placed in culture. In this case, helper cells responded to a for-eign MHC class II antigen, but cytotoxic T cells were able to lyse target cells carry-ing the MHC class I molecules of the stim-ulating (genetically different individual) cells. The in vivo reactions between indi-viduals undergoing transplantation will be discussed in more detail.
In contrast, delayed-type hypersensi-tivity reactions are mediated by specific T cells that produce TH1-type cytokines upon exposure to antigen. An example of this type of reaction is the PPD reaction, or tuberculin test. When the antigen is injected under the skin of an individual who was previously infected with Mycobacterium tuberculosis, a reaction in the skin evolves over 48 to 72 hours in which there is local swelling and induration >10 mm. If the site is biopsied, one finds a T-cell and macro-phage infiltration. Injection of the same material in a noninfected individual pro-duces little or no induration, and the his-tology is essentially negative. Whereas the cells in this case do not kill the organism,
most individuals infected surround the organism in a caseous inflammatory lesion, which does not allow the organism to spread. The in vivo state of the lesions will be discussed in more detail.
Nonspecific Effector Molecules
There are a number of nonspecific mol-ecules that affect the immune response, especially antibody production. These major factors are as follows: phagocytic cells such as neutrophils and macrophages, which remove antigens and bacteria, and complement, which can either destroy the organism or facilitate its destruction. The role of many of these factors will be discussed in more detail in later, but a brief outline of their functions is war-ranted here.
COMPLEMENT
The complement component system consists of a series of heat-liable proteins, and they normally exist as inactive precur-sors. However, once activated each compo-nent may act as an enzyme and cleaves the next component in the sequence.
Each precursor is cleaved into two or more components, and the major frag-ment (designated “b”) has two biologically active sites. One is for binding to cell mem-branes and the other is for enzymatic cleav-age of the next component. The control of the sequence relies on either spontaneous decay or specific inactivation of these com-ponents. Minor fragments play a role in the fluid phase, acting as chemotactins.
The major function of the complement system is to help in the opsonization of micro-organisms and immune complexes. These components plus antibody are more read-ily recognized by macrophages and more
readily bound and phagocytosed through IgG: Fc and C3b receptors. Immune com-plexes are handled in a similar fashion, activating the classical pathway comple-ments. Individuals who lack one of the classical pathway components are prone to immune complex disease.
The minor complement fragments con-tribute to the immune response by activating the inflammatory response. For example, some increase vascular permeability (C3a); others are chemotactins for neutrophils and macrophages (C5a) and not only promote leucocytosis in the bone marrow but attract these cells to the site of inflammation.
The critical step in complement activa-tion is the cleavage of the C3 component by complement-derived enzymes called C3 convertases. This results in the presence of C3b, which mediates a number of vital biological activities. The cleavage of C3b can be initiated by three routes (classical, alternative, and lectin), but each route is in response to different stimuli. Individu-als who are deficient in C3 are obviously predisposed to bacterial infections and immune complex disease.
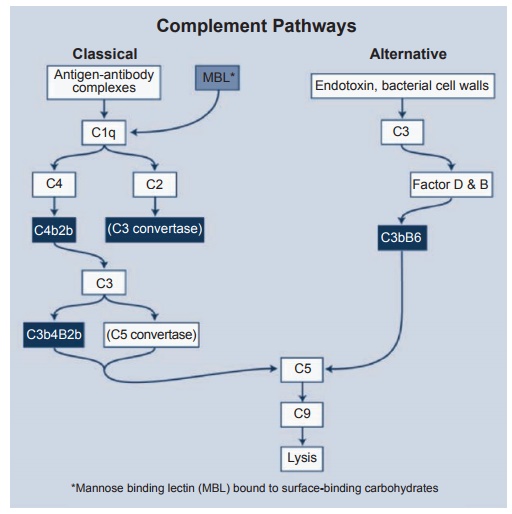
Each of these routes will be examined in more detail (see Figure 1.9).
1. Classical pathway: As its name implies, this is the usual pathway whereby anti-gen–antibody complexes in the presence of complement destroy the invading organism. The antibody (either IgM or IgG) causes a conformational change in the Fc portion of the antibody to reveal a binding site for the first component of complement C1q. This component consists of six subunits and reacts with the Fc via its globular heads. The acti-vation of this component requires the binding of two globular heads for acti-vation. Thus, one molecule of IgM with easily activate C1q, while the ability of IgG, which has only two sites to activate C1q, is low. IgA, IgD, and IgE do not activate the classical pathway. C1q in turn activates C4 and C2, generating the complex C4b2b, which is the C3 “con-vertase” of the classical pathway. After the splitting of C3 into C3a and C3b is achieved, C3a possesses anaphylatoid and chemotactic activity. However, more important is C3b, which forms the complex C3b4b2b, which is the C5 convertase and initiates the final “lytic” attack complex.
2. The alternative pathway is phyloge-netically older than the classical but was not generally accepted until the 1960s. Again, the control reaction is the activation of C3. In contrast to the clas-sical pathway, however, this pathway bypasses antibody, C1, C4, and C2, and it is bacterial cell walls or endotoxin that activates C3. C3b here is unstable and, if an appropriate receptor is not found, it decays and the molecule becomes inactive. However, if a receptor surface is present, then the C3b molecules bind and remain active. Then C3b can use factors D and B of the alternative path-way to form the active enzyme C3bBb; this complex then becomes stabilized in the presence of properdin.
This molecule then can chose between two pathways. It can break down more C2, providing more C3b. Or it becomes stabilized to form the C5 convertase of the alternative pathway.
3. Lectin pathway: The third pathway of complement activation is created by the mannose-binding lectin MBL, a circulating protein that binds to carbo-hydrate on the surface of many micro-organisms. MBL (structurally related to C1q) activates complement through a serine protease known as MBL-associated serine protease. Deficiencies in circulating levels of MBL are asso-ciated with frequent infections in childhood.
Once these components are activated, that is, C3b, 4b2b or C3bBb, and proper-din, these molecules trigger sequentially C5, C6, C7, C8, and C9, which leads to the final lytic pathway and lysis of the target cell. The target can be a red cell, a virally infected cell, or a bacterium. Electron microscopy has shown that this complex binds to the cell membrane and actually punches a hole in the cell. Salts and water pass through the hole and the water fills the cell, eventually leading to swelling and destruction of the cell.
The control of the complement activa-tion is important since many of its compo-nents induce inflammation. This control is executed in the following ways. First, many of the activated components are unstable and will decay rapidly if the next sequence is not present. Second, there are specific inhibitors of each component, such as C1 esterase, which inhibits factors I and H. Finally, the cells themselves contain pro-teins that increase the rates of breakdown of these products.
In summary, all acute phase comple-ment components are acute phase proteins and the rate of increase occurs shortly after injury or infection. As will be seen later, there is considerable interaction between the complement system and other pathways such as clotting, fibrinolytic, and kimin pathways.
Related Topics