Chapter: Modern Analytical Chemistry: Evaluating Analytical Data
Analytical Data: Characterizing Experimental Errors
Characterizing Experimental Errors
Realizing that our data for the mass of a penny can be characterized by a measure
of central tendency and a measure
of spread suggests
two questions. First,
does our measure of central tendency
agree with the true, or expected value?
Second, why are our
data scattered around
the central value?
Errors associated with
central tendency reflect the accuracy of the analysis, but the precision of the analysis
is determined by those
errors associated with the spread.
Accuracy
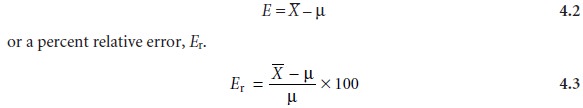
Although the mean is used as the measure of central tendency
in equations 4.2 and
4.3, the median could also be used.
Errors affecting the
accuracy of an analysis are
called determinate and
are char- acterized by a systematic deviation from the
true value; that
is, all the
individual measurements are either too large or too small. A positive determinate error results in a central value
that is larger
than the true
value, and a negative determinate error leads to a central value that is smaller than the true value. Both positive and nega-
tive determinate errors
may affect the
result of an analysis, with
their cumulative ef- fect leading to a net positive or negative determinate error. It is possible, although not likely, that positive and negative determinate errors may be equal, resulting in a central value with no net determinate error.
Determinate errors may be divided into four categories: sampling errors,
method errors, measurement errors, and personal
errors.
Sampling Errors
We introduce determinate sampling errors
when our sampling strategy fails to provide
a representative sample.
This is especially important when sampling heterogeneous materials. For example, determining the environmental
quality of a lake by sampling a single location
near a point source of pollution, such as
an outlet for industrial effluent, gives misleading results.
In determining the mass
of a U.S. penny, the strategy for selecting pennies
must ensure that pennies from other countries are not
inadvertently included in the sample.
Method Errors
Determinate method
errors are introduced when assumptions about the relationship between
the signal and the analyte
are invalid. In terms of the
general relationships between
the measured signal
and the amount
of analyte

method errors exist
when the sensitivity, k, and the signal
due to the reagent blank, Sreag, are incorrectly determined. For example,
methods in which
Smeas is the mass of
a precipitate containing the analyte
(gravimetric method) assume that the sensitiv-
ity is defined by a pure precipitate of known stoichiometry. When this assumption fails, a determinate error
will exist. Method
errors involving sensitivity are mini- mized by standardizing the method, whereas
method errors due to interferents
present in reagents are minimized by using a proper reagent
blank. Method errors due to interferents in the sample cannot be minimized by a reagent
blank. Instead, such interferents must be sepa- rated from the analyte
or their concentrations determined independently.
Measurement Errors
Analytical instruments and equipment, such as glassware and balances, are usually
supplied by the manufacturer with a statement of the item’s maximum measurement error, or tolerance.
For example, a 25-mL volumetric flask might have a maximum
error of ±0.03
mL, meaning that the actual
volume contained by the flask lies within the range of 24.97–25.03 mL. Although expressed as a range, the
error is determinate; thus, the flask’s
true volume is a fixed
value within the stated
range. A summary
of typical measurement errors for a variety of analytical equipment is given
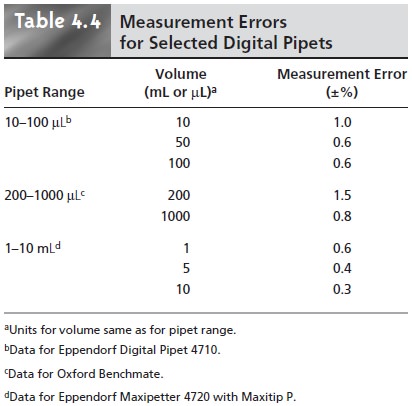
in Tables 4.2–4.4.
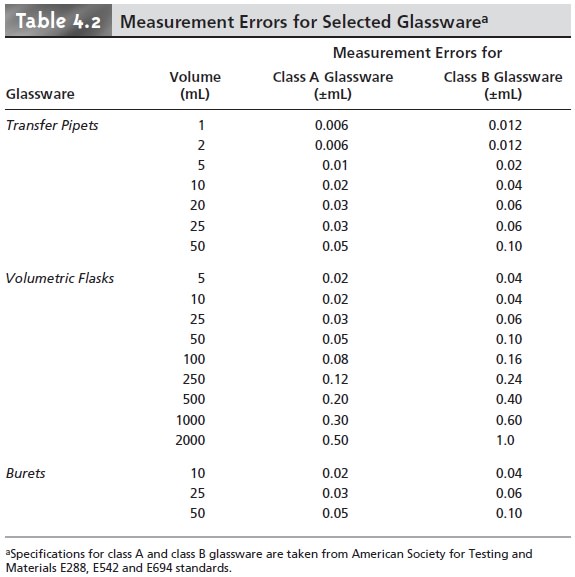
Volumetric glassware is categorized by class. Class
A glassware is manufactured
to comply with tolerances specified by agencies such as the National Institute of Standards and Technology. Tolerance levels for class A glassware are small enough that such glassware normally
can be used without calibration. The tolerance levels for class B glassware are usually twice
those for class
A glassware. Other
types of vol- umetric glassware, such as beakers
and graduated cylinders, are unsuitable for
accu- rately measuring volumes.
Determinate measurement errors can be minimized by calibration. A pipet can be
calibrated, for example,
by determining the mass of water that it delivers
and using the density
of water to calculate the actual volume
delivered by the pipet. Al- though glassware and instrumentation can be calibrated, it is never
safe to assume that the calibration will remain unchanged during an analysis. Many instruments, in particular, drift out of calibration over time. This complication can be minimized by frequent recalibration.
Personal Errors
Finally, analytical work is always
subject to a variety of personal errors, which can include
the ability to see a change in the color of an indicator
used to signal the end point of a titration; biases, such as consistently overestimat- ing or underestimating the value on an instrument’s readout scale; failing
to cali- brate glassware and
instrumentation; and misinterpreting procedural directions. Personal errors
can be minimized with proper
care.
Identifying Determinate Errors
Determinate
errors can be difficult to detect.
Without knowing the true value for an analysis, the usual situation
in any analysis with meaning, there
is no accepted value with which the experimental result
can be compared. Nevertheless, a few strategies can be used to discover
the presence of a
determinate error.
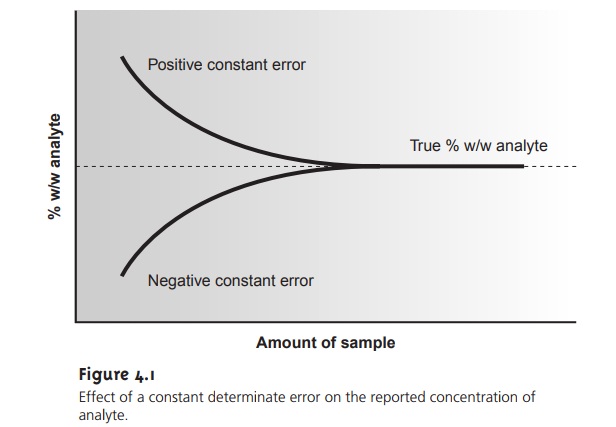
Some determinate errors can be detected
experimentally by analyzing several samples of different size. The magnitude of a constant determinate error is the same for all samples
and, therefore, is more significant when analyzing smaller
sam- ples. The presence
of a constant determinate error can be detected by running sev- eral
analyses using different amounts of sample,
and looking for a systematic change in the property being measured. For example, consider a
quantitative analysis in which we separate the analyte from
its matrix and
determine the analyte’s mass. Let’s assume that the sample
is 50.0% w/w analyte; thus,
if we analyze a 0.100-g sample, the analyte’s true mass is 0.050 g. The first
two columns of Table 4.5 give
the true mass of analyte
for several additional samples. If the analysis has a positive constant determinate error of 0.010 g, then the experimentally determined mass for
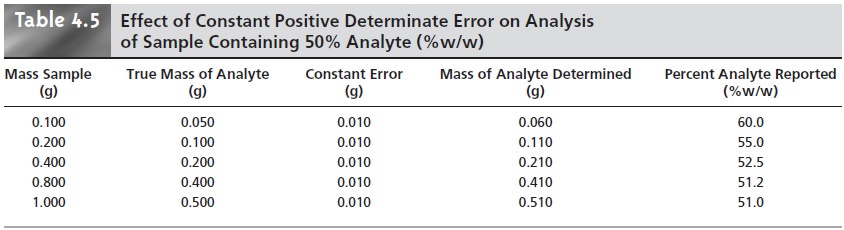
any sample will
always be 0.010
g, larger than
its true mass
(column four of Table
4.5). The analyte’s reported weight
percent, which is shown in the last column of Table
4.5, becomes larger
when we analyze
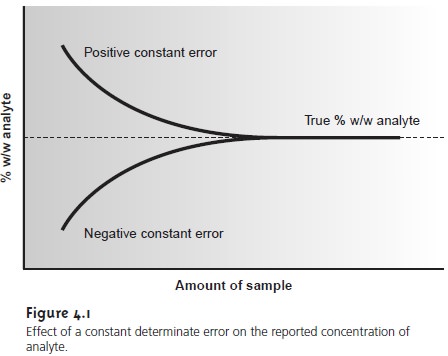
smaller samples. A graph of % w/w
ana- lyte versus amount of sample shows a distinct upward trend for small amounts
of sample (Figure 4.1). A smaller
concentration of analyte
is obtained when analyzing
smaller samples in the presence of a constant negative determinate error.
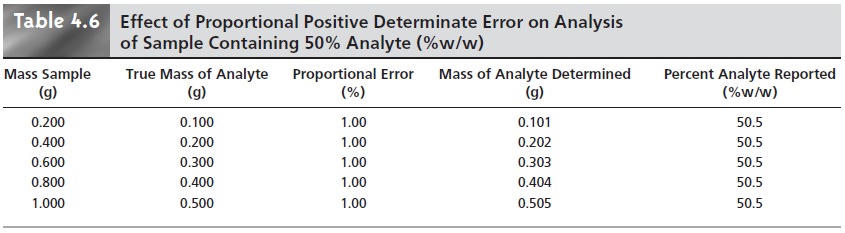
A proportional determinate error, in
which the error’s
magnitude depends on the
amount of sample,
is more difficult to detect since
the result of an analysis
is in- dependent of the amount of sample.
Table 4.6 outlines
an example showing
the ef- fect of a positive proportional error of 1.0% on the analysis of a sample
that is 50.0% w/w in analyte.
In terms of equations 4.4 and 4.5, the reagent
blank, Sreag, is an example
of a constant determinate error,
and the sensitivity, k, may be affected by proportional errors.
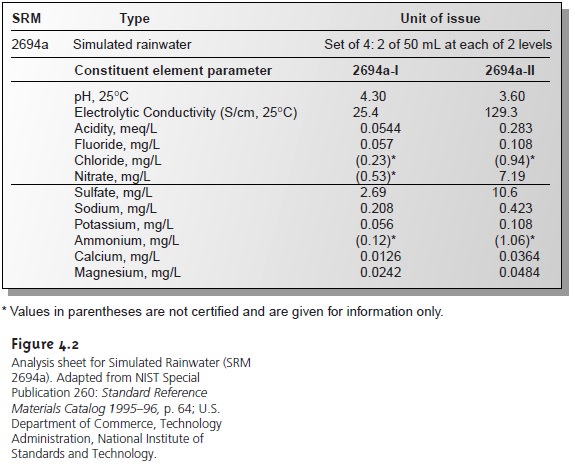
Potential determinate errors
also can be identified by analyzing a standard sam- ple
containing a known
amount of analyte
in a matrix similar to that of the samples being analyzed. Standard samples
are available from
a variety of sources, such
as the National Institute of Standards and Technology (where
they are called
standard reference materials) or the American
Society for Testing
and Materials. For exam-
ple, Figure 4.2 shows an analysis sheet
for a typical reference material. Alternatively, the sample can
be analyzed by an independent
method known to give accurate
results, and the re-
sults of the two methods
can be compared. Once identified, the source
of a determinate error can be
corrected. The best
prevention against errors
affect- ing accuracy, however,
is a well-designed procedure
that identifies likely sources of determinate errors, coupled with careful laboratory work.
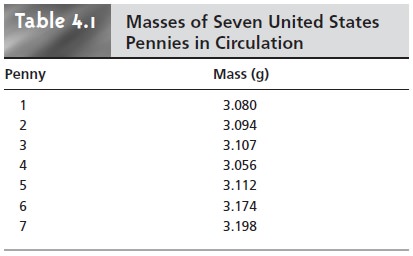
The data in Table 4.1 were obtained using a calibrated balance, certified by the manufacturer to have a tolerance of less than ±0.002 g. Suppose the Treasury Department reports that the mass of a1998 U.S. penny is approximately 2.5 g. Since the mass of every penny in Table 4.1 exceeds the re- ported mass by an amount significantly greater than the balance’s tolerance, we can safely conclude that the error in this analysis is not due to equip- ment error.
Precision
Precision is a measure of the spread
of data about
a central value
and may be ex-
pressed as the range, the standard deviation, or the variance. Precision is commonly divided into two categories:
repeatability and reproducibility. Repeatability
is the precision obtained when all measurements are made by the same analyst during
a single period of laboratory work,
using the same solutions and equipment. Repro-
ducibility, on the
other hand, is the precision obtained under any
other set of con-
ditions, including that between analysts, or between laboratory sessions for a single
analyst. Since reproducibility includes additional sources
of variability, the repro-
ducibility of an analysis can
be no better than its
repeatability.
Errors affecting the distribution of measurements around a central
value are called indeterminate
and are characterized by a random variation in both magni- tude and direction. Indeterminate errors
need not affect
the accuracy of an analy- sis. Since indeterminate errors
are randomly scattered around a central
value, posi- tive and
negative errors tend
to cancel, provided that enough measurements are made. In such situations the mean or median is largely unaffected by the precision of the analysis.
Sources of Indeterminate Error
Indeterminate errors can be traced
to several sources, including the collection of samples, the manipulation of
samples during the analysis, and the making of measurements.
When collecting a sample, for instance, only a small
portion of the available
material is taken, increasing the likelihood that small-scale inhomogeneities in the sample will affect the repeatability of the analysis. Individual pennies, for example,
are expected to show variation
from several sources,
including the manufacturing process, and the loss of small amounts of metal or the addition
of dirt during circu- lation. These
variations are sources
of indeterminate error
associated with the sam-
pling process.
During the analysis
numerous opportunities arise
for random variations in the way individual samples are treated.
In determining the mass of a penny,
for exam- ple, each penny should
be handled in the same manner. Cleaning
some pennies but not
cleaning others introduces an indeterminate error.
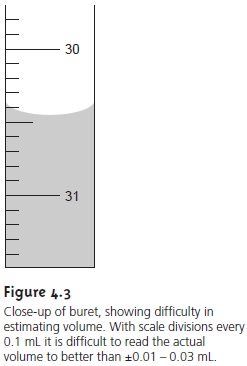
Finally, any measuring device
is subject to an indeterminate error in reading
its scale, with the last digit
always being an estimate subject
to random fluctuations, or background noise. For example, a buret with scale divisions every 0.1 mL has an in-
herent indeterminate error
of ±0.01 – 0.03 mL when estimating the volume to the
hundredth of a milliliter (Figure
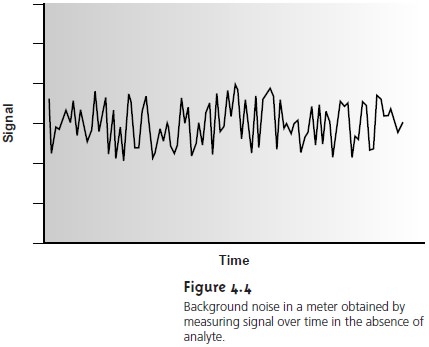
4.3). Background noise
in an electrical meter (Figure 4.4) can be evaluated by recording the signal without
analyte and observing
the fluctuations in the signal over time.
Evaluating Indeterminate Error
Although it is impossible to eliminate indetermi- nate error, its effect can be minimized if the sources
and relative magnitudes of the indeterminate
error are known. Indeterminate errors may be estimated by an ap- propriate measure of spread.
Typically, a standard deviation is used, although in some cases
estimated values are
used. The contribution from analytical instruments and equipment are easily
measured or estimated. Inde- terminate errors
introduced by the
analyst, such as in-
consistencies in the treatment of individual samples, are more difficult to estimate.
To evaluate the effect of indeterminate error
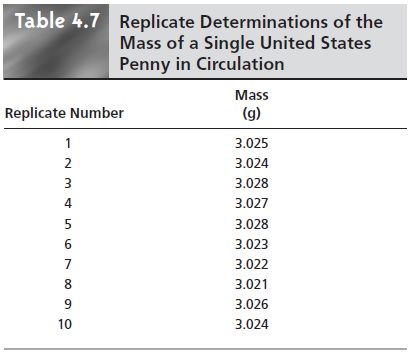
on the data in Table 4.1,
ten replicate determinations of the mass of a single penny were made, with results shown in Table 4.7. The standard
deviation for the data in Table 4.1 is 0.051,
and it is 0.0024 for the data in Table 4.7. The significantly better precision when determining the mass of a single penny sug- gests
that the precision of this analysis
is not limited by the balance
used to measure
mass, but is due to a
significant variability in the masses of individual
pennies.
Error and Uncertainty
Analytical chemists make a distinction between error and uncertainty.3 Error is the difference between a single measurement or result and its true value. In other words, error is a measure of bias. As discussed earlier,
error can be divided into de-
terminate and indeterminate sources. Although we can correct
for determinate error, the
indeterminate portion of the error
remains. Statistical significance testing, provides a way to determine whether a bias resulting from determinate error
might be present.
Uncertainty expresses the
range of possible values that a measurement or result
might reasonably be expected to have. Note that this definition of uncertainty is not
the same as that for precision. The precision of an analysis, whether reported as a
range or a standard deviation, is calculated from
experimental data and
provides an estimation of indeterminate error
affecting measurements. Uncertainty accounts for all errors,
both determinate and
indeterminate, that might
affect our result.
Al- though we always
try to correct
determinate errors, the
correction itself is subject to random effects or indeterminate errors.
To illustrate the difference between
precision and un- certainty, consider the use of a class A 10-mL pipet for de- livering solutions. A pipet’s
uncertainty is the range of volumes in which its true volume
is expected to lie. Sup- pose you purchase a 10-mL class
A pipet from
a labora- tory supply
company and use it without
calibration. The pipet’s tolerance value of ±0.02
mL (see Table
4.2) repre- sents your uncertainty since your best estimate of its vol- ume
is 10.00 mL ±0.02 mL.
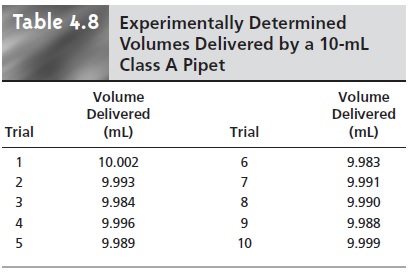
Precision is determined ex- perimentally by using the pipet several times, measuring
the
volume of solution delivered each time. Table 4.8 shows results
for ten such trials that have a mean of 9.992
mL and a standard deviation of 0.006. This standard devi- ation represents the precision
with which we expect to be
able to deliver a given
solution using any class A 10-mL
pipet. In this case the uncertainty in using a pipet is worse
than its precision. Interestingly, the data in Table 4.8 allow us to calibrate this specific pipet’s
delivery volume as 9.992 mL.
If we use this vol- ume as a better estimate of this pipet’s
true volume, then the uncertainty is ±0.006. As expected, calibrating the pipet
allows us to lower its uncertainty.
Related Topics