Chapter: Plant Biochemistry: Biotechnology alters plants to meet requirements of agriculture, nutrition and industry
A gene is isolated
A gene is isolated
Let us consider the case where a transgenic plant A is to be generated, which synthesizes a foreign protein (e.g., a protein from plant B). For this, the gene encoding the corresponding protein first has to be isolated from plant B. Since a plant contains between 25,000 and 50,000 structural genes, it will be difficult to isolate a single gene from this very large number.
A gene library is required for the isolation of a gene
To isolate a particular gene from the great number of genes existing in the plant genome, it is advantageous to make these genes available in the form of a gene DNA library. Two different kinds of gene libraries can be prepared.
To prepare a genomic DNA library, the total genome of the organism is cleaved by restriction endonucleases into fragments of about 15 to several 100 kbp. Digestion of the genome in this way results in a very large number of DNA fragments, which frequently contain only parts of genes. These fragments are inserted into a vector (e.g., a plasmid or a bacteri-ophage) and then each fragment is amplified by cloning, usually in bacteria.
To prepare a cDNA library, the mRNA molecules present in a spe-cific plant tissue are first isolated and then transcribed into corresponding cDNAs by reverse transcriptase. The mRNA is isolated from a tissue in which the corresponding gene is expressed at high levels. The cDNAs are inserted into a vector and amplified by cloning. In contrast to the fragments of the genomic library, the resulting cDNAs contain no introns and can therefore, after transformation, be expressed in prokaryo-tes to synthesize the foreign proteins. Since a cDNA contains no promoter regions, such an expression requires a prokaryotic promoter to be added to the cDNA.
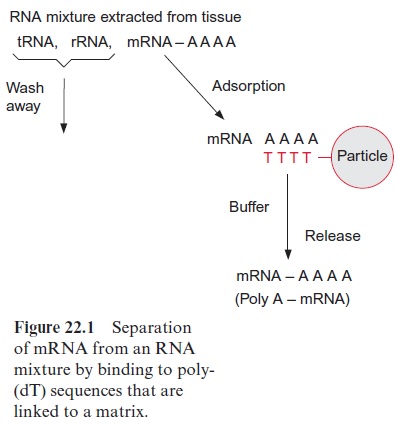
To prepare a cDNA library from leaf tissue, for example, the total RNA is isolated from the leaves, of which the mRNA may amount to only 2%. To separate the mRNA from the bulk of the other RNA species (rRNAs and tRNAs), one makes use of the fact that eukaryotic mRNA contains a poly(A) tail at the 3 terminus (see Fig. 20.8). This allows mRNA to be separated from the other RNAs by affinity chromatography. The column material consists of solid particles of cellulose or another matrix to which a poly-deoxythymidine oligonucleotide (poly-dT) is linked. When an RNA mixture extracted from leaves is applied to the column, the poly-(A) tails of the mRNA hybridizes to the poly-(dT) of the column, whereas other RNAs do not bind and run through (Fig. 22.1). With an appropriate buffer, the bound mRNA is eluted from the column.
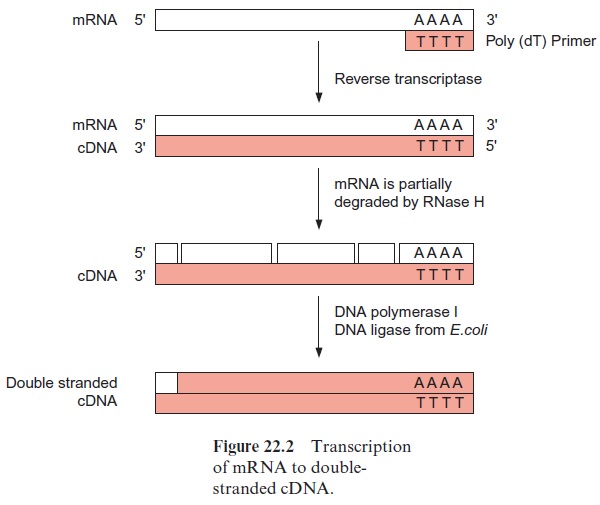
The poly-A enriched RNA preparation is then used to synthesize by reverse transcriptase a cDNA strand complementary to the mRNA. The reverse transcriptase starts its synthesis at a poly-(dT) primer (Fig. 22.2). Subsequently, the mRNA is hydrolyzed by a ribonuclease either completely or, as shown in the figure, only partly. The latter method has the advan-tage that the mRNA fragments can serve as primers for the synthesis of the second cDNA strand by DNA polymerase. DNA polymerase I replaces mRNA fragments successively by DNA fragments and these are linked to each other by DNA ligase. A short RNA section remains, which is not replaced at the end of the second cDNA strand, but this is of minor impor-tance, since in most cases the mRNA at the 5 terminus does not encode protein sequence (see Fig. 20.8).
The double-stranded (ds) cDNA molecules thus formed from the mRNA molecules are amplified by cloning. Plasmids or bacteriophages can be used as cloning vectors. Nowadays a large variety of made-to-measure phages and plasmids are commercially available for many special purposes. A distinc-tion is made between vectors that amplify only DNA andexpression vectors by which the proteins encoded by the amplified genes can be synthesized.
A gene library can be kept in phages
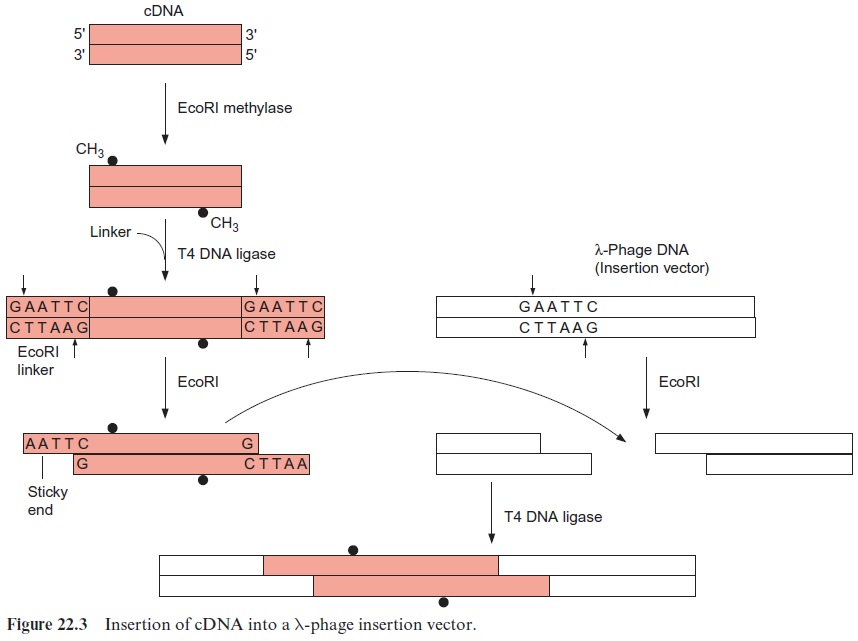
Figure 22.3 shows the insertion of cDNA into the DNA of a l phage. In the example shown here, the phage DNA possesses a cleavage site for the restriction endonuclease EcoRI. The cDNA double strand is first methylated by an EcoRI methylase at its own EcoRI restriction sites in order to protect this restriction site within the cDNA. DNA ligase is then used to link chemically synthesized double-stranded oligonucleotides with an inbuilt restriction site (in this case for EcoRI) to both ends of the double-stranded cDNA. These oligonucleotides are called linkers. The restriction endonuclease EcoRI cleaves this linker as well as the phage DNA and thus generates sticky ends at which the complementary nucle-otides of the cDNA and the phage DNA can anneal by base pairing. The DNA strands are then linked by DNA ligase, and in this way the cDNA is inserted into the phage vector.
The phage DNA with the inserted cDNA is packed in vitro into a phage protein coat (Fig. 22.4), using a packing extract from phage-infected bacte-ria. In this way one obtains a gene library, in which the cDNA from many different mRNAs of the leaf tissue are packed in phages. After infecting bacteria they can be amplified ad libitum, whereby each packed cDNA is one individual clone harboring one plant gene.
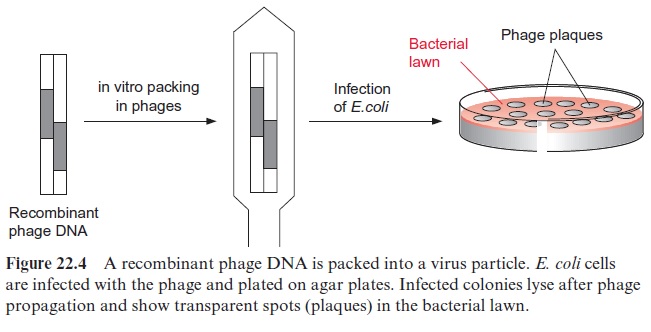
The bacteria are infected by mixing them with the phages. At first bacte-ria grow on the agar plates to produce a bacterial lawn, and then the phages, which have been priorly propagated in bacteria, are added on top. After incubation, the infected and lysed bacterial colonies appear on the agar plate as clear spots within the bacterial lawn and are called plaques. These plaques contain newly formed phages, which can be multiplied further. It is custom-ary to plate a typical cDNA gene library on about 10 to 20 agar plates of ca. 20 cm diameter. Ideally, each of these plaques contains only one clone. From these plaques, the phage clone containing the cDNA of the desired gene is selected, using specific probes as will be described later.
A gene library can also be propagated in plasmids
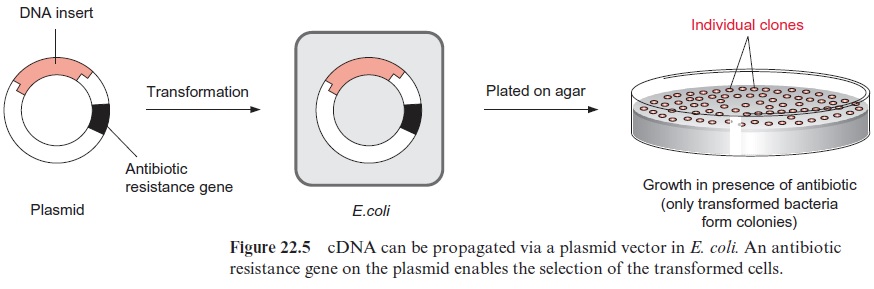
cDNA cloning into plasmids is carried out similarly to that described via restriction. To clone a cDNA gene library into plasmids, cDNA is inserted into plasmids via a restriction cleavage site in more or less the same way as in the insertion into phage DNA (Fig. 22.5). The plasmids are then trans-ferred into E. coli cells. The transfer can be brought about by treating the cells with CaCl2 to make their membrane more permeable (competent) to the plasmid. The cells are then mixed with plasmid DNA and exposed to a short heat shock. In order to select the transformed bacterial cells from the large majority of untransformed cells, the plasmid vector of the trans-formed cells is provided with a marker. The plasmid vector contains anantibiotic resistance gene, which renders the bacteria resistant to growing on a certain antibiotic, such as ampicillin or tetracycline. When the corre-sponding antibiotic is added to the culture medium, only cells containing the plasmid survive and grow, whereas the other nontransformed cells die. After plating on an agar culture medium, bacterial colonies develop which and can be recognized as spots.
In order to verify that a plasmid actually contains an inserted DNA sequence (insert), the blue/white screen can be used. Specific vectors which encode the enzyme β-galactosidase were designed (Fig. 22.6). This enzyme hydrolyzes the colorless compound X-Gal into an insoluble blue product. When X-Gal is added to the agar culture medium, all the clones that do not contain a DNA insert, and therefore contain an intact β–galactosidase gene, form blue colonies. If a DNA fragment is inserted into the cleavage site of the β -galactosidase gene, this gene is interrupted and no longer able to encode a functionalβ-galactosidase. Therefore the corresponding colo-nies are not stained blue but remain white (blue/white selection).

A gene library is screened for a certain gene
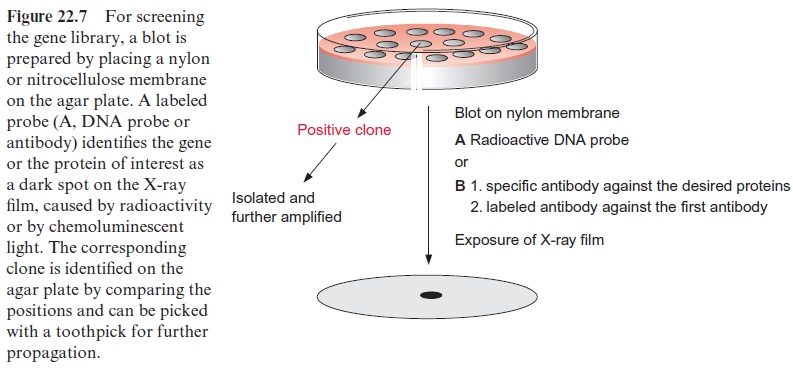
Specific probes that correspond to the gene of interest are employed to screen the bacterial colonies or phage plaques. A blot is made by placing a nylon or nitrocellulose membrane on top of the bacterial colonies or phage plaques (Fig. 22.7). Some of the phages of the plaques or the bacterial clone will bind to the blotting membrane, although most of them remain on the agar plate. After special treatment to lyse bacteria or phages the DNA of the bacteria or phages is fixed to the membrane. Two kinds of probes can be used to screen the DNA or protein bound to the blotting membrane derived from the phages or bacteria:
1. Specific DNA probes to label the DNA of the desired clone by hybridi-zation and
2. Specific antibodies to identify the desired protein expressed as gene product of the clone (Western blot).
A clone is identified by antibodies which specifically detect the gene product
Antibodies against a certain protein are often used to identify the correspond-ing gene (e.g., by Western blot). This method is only applicable when approxi-mately 1 mg of the protein of the gene of interest is purified beforehand in order to obtainpolyclonal antibodies by immunization of animals. Furthermore, the cDNA library must be inserted in an appropriate expression vector, so that transcription and translation of the foreign proteins can occur in E. coli. The vector contains a promoter sequence, which controls initiation of transcription of the inserted gene, and often also a sequence for termination of the transcrip-tion at its end.
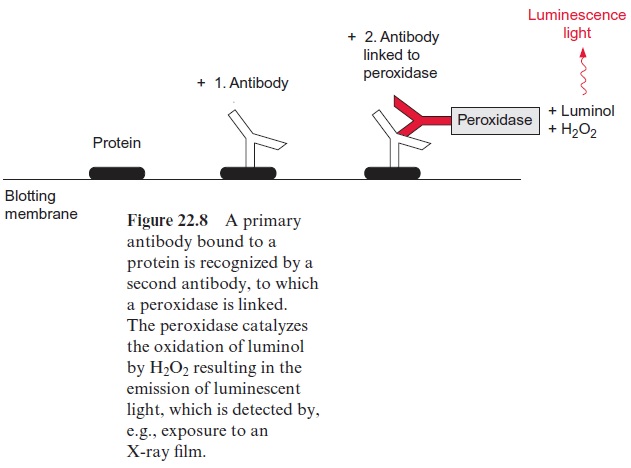
In practice, bacteria bound to the blotting membrane are disrupted by alkali treatment and the released bacterial proteins are fixed to the membrane (Fig. 22.7). When phages are used as vectors, cell disruption is not required, since the phages themselves lyse the bacterial cell and thus liberate the cel-lular proteins, which are then fixed to the blotting membrane. Afterwards, antibodies are added, which bind specifically to the corresponding protein, but are washed off from all other parts of the membrane. Usually a second antibody, which recognizes the first antibody, is used to detect the first bound antibody (Fig. 22.8). Formerly the second antibody was labeled with radio-active 125iodine. Nowadays more often theECL-technique (enhanced lumi-nescence) is employed. This entails a peroxidase from radish being attached to the second antibody. This peroxidase catalyzes the oxidation of added luminol (3-aminophthalic acid hydrazide) with H2O2 as oxidant. This reac-tion is accompanied by the emission of blue luminescent light, which can be detected after about 1 hour exposure to an X-ray film. Using a lumines-cence enhancer, the intensity of this chemoluminescence can be increased by a factor of 1,000. A positive colony can be recognized as a dark spot in the autoradiography (Fig. 22.7). The position of the positive clone on the agar plate can be identified from the position of the spot on the blotting mem-brane. After the first screening, due to a high density of the plated bacteria an apparently positive clone may actually contain several clones. For this reason, the colony, picked up from the positive region with a toothpick, is diluted and plated again on an agar plate. By repeating the screening pro-cedure described above (rescreening), finally single pure clones are obtained. Positive phage plaques are also regrown in bacteria and plated again in order to obtain pure clones.
A clone can also be identified by DNA probes
In this procedure, the phages or bacteria present on the blotting mem-brane are first lysed and the proteins are removed. The remaining DNA is then denatured to obtain single strands, which are tightly bound to the membrane. Complementary DNA sequences, which are radioactively labeled by 32P-labeled deoxynucleotides or by digoxigenin-labeled dUTP (DIG11 dUTP), are used as probes. These probes bind to complementary DNA sequences present on the blotting membrane by hybridization. The identification of the positive clones proceeds via autoradiography or with digoxigenin antibodies that have been conjugated with a chemilumines-cence dye.
Chemically synthesized oligonucleotides of about 20 bases are also employed as DNA probes. These probes are radioactively labeled at the 5 end by 32P-phosphate and are used particularly when only low quantities of the purified proteins are available, and which are not sufficient for the gen-eration of antibodies. For the synthesis of oligonucleotides with automatic synthesizers some sequence information of the gene of interest is needed, either from homologous genes of other species or a partial amino acid sequence of the corresponding protein. With very low amounts of protein, it is possible to determine part of the N-terminal amino acid sequence of the protein by micro-sequencing. From such a partial amino acid sequence, the corresponding DNA sequence can be deduced according to the universal genetic code. The corresponding oligonucleotide probes are subsequently produced by chemical synthesis using automatic synthesizers. However, the degeneracy of the genetic code implies that amino acids are often encoded by more than one nucleotide triplet (Fig. 22.9). This is taken into account during the design of oligonucleotides. When, for example, the third base of the triplet encoding lysine can be either an A or a G, a mixture of both nucleotides is added to the synthesizer during the reaction with the third nucleotide of this triplet. In order to introduce the third nucleotide of the triplet encoding alanine triplet, a mixture of all four nucleotides is added to the synthesizer. The synthesized “degenerate” oligonucleotide shown in Figure 22.9 isthus, in fact, a mixture of 48 different oligonucleotides, only one of which contains the correct sequence of the desired gene.

When the protein encoded by the desired gene could not be purified, a corresponding gene section from related organisms can be sometimes used as a homologous probe. Domains with specific amino acid sequences are often conserved in enzymes or other proteins (e.g., translocators) even from distantly related plants, and are encoded by correspondingly similar gene sequences.
After the DNA for the desired protein has been successfully isolated, usually the next step is to determine the complete nucleotide sequence, mostly by automatic analyzers, which will not be described here.
Genes encoding unknown proteins can be functionally assigned by complementation
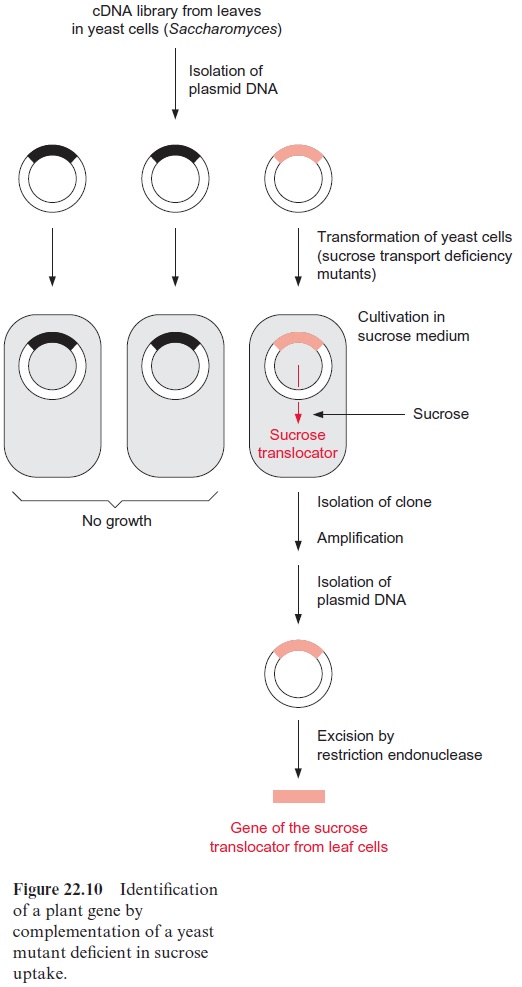
In some cases it has been possible to isolate the cDNA encoding an unknown protein. One method for identifying the function of the gene is by the complementation of deficiency mutants of bacteria or yeast. To do so, the plasmids from a cDNA library are transformed into bacteria or yeast mutants (Fig. 22.10). Plasmids that can be amplified in E. coli as well as in yeast are available as cloning vectors and can be used to express the encoded protein. Several plant translocators, including the sucrose trans-locator involved in phloem loading, have been identified by complementation. To identify the gene of the sucrose translocator, a yeast deficiency mutant was employed that had lost the ability to take up sucrose and therefore could no longer use sucrose as a nutritional source. This mutant was transformed with plasmids from a plant cDNA library, where the plasmid DNA was provided with a yeast promoter in order to express the inserted DNA within the yeast cell. After plating the transformed yeast cells on a culture medium with sucrose as the only carbon source, a yeast clone was found that grew on the sucrose cultivation medium. This indi-cated that transformation with the corresponding plasmid from the plant cDNA library had generated yeast cells that produced the plant sucrose translocator and incorporated it into their cell membrane. After this positive yeast clone had been amplified, plasmid cDNA was isolated and sequenced. The cDNA sequence yielded the amino acid sequence of the previously unknown sucrose translocator.
Genes can be identified with the help of transposons or T-DNA
Another possibility for identifying genes, aside from the function of their gene products, is to use transposons. Transposons are DNA sequences that can “jump” within the genome of a plant, which sometimes results in a recognizable elimination of a gene function. This could, for example, be the loss of the ability to synthesize a flower pigment. In such a case, a gene probe based on the known transposon sequence is used to identify by DNA hybridization the region of the genome in which the transposon has been inserted. By using the cloning and screening proce-dures already mentioned, it is possible to identify a gene, in our example a gene for the enzyme of flower pigment synthesis, and to determine the amino acid sequence of the corresponding enzyme by DNA sequence anal-ysis. Labeling a gene with an inserted transposon is calledgene tagging.
An alternative method, which has superseded the transposon tech-nique of tracking genes, is the T-DNA insertion technique, which will be described in the next section in detail. Inserting T-DNA into the genome causes random mutations (T-DNA-insertion mutants). The gene locus into which the T-DNA has “jumped” is identified by using a gene probe and is subsequently sequenced. The complete sequence of the mutated gene is then identified, usually by comparing the relevant gene region with a data-base of the known sequence of T-DNA insertion mutants. For instance, the firm Syngenta provides a data bank with the sequences of about 100,000 T-DNA insertion mutants of Arabidopsis.
Related Topics