Chapter: Essential Clinical Immunology: Immunological Aspects of Infection
Viral Infections
VIRAL INFECTIONS
The clinical spectrum of viral disease is wide, and because there is such variation, we will use the template of herpes viruses as an example. In general, viral infections are self-limited and usually produce long-term immunity, and secondary attacks by the same virus are uncommon. Of the large number of herpes viruses, only eight infect humans, and each will be discussed in some detail. However, two general features of pathogens of these viruses are important. First, there must be close contact between infected and noninfected individuals for transmission to occur and no intermediate host is involved. Second, after the primary infection, herpes virus will persist in the host for life.
To eliminate virions from entering noninfected cells and to eliminate virus-infected cells, two major pathways of the immune response are initiated. The humoral response is primarily directed against virions, while the T-cell response is primarily directed against infected cells. The humoral response may directly neutralize the virus, but complement-dependent enhancement of viral phago-cytosis or complement lysis of virus also may occur.
Epstein-Barr Infection
Infectious mononucleosis is caused by the Epstein-Barr virus (EBV), and in devel-oping countries, 99 percent of children demonstrate antibodies to the virus by age 3. In developed countries, the age is much later and clinical infection usually occurs in the age group of 15 to 25 years. The virus is excreted in the oropharyngeal fluids for some months after the infection and is responsible for person-to-person transmission.
Hoagland observed that a female col-lege student traveling from New York to Boston met several different male students on the train and exchanged kisses with all of them. All of these contacts came down with infectious mononucleosis, which is often referred to as the “kissing disease.”
The pattern of antibody response to different EBV antigens helps distinguish acute or subclinical infections from postin-fections with the virus. IgM antibodies to viral capsid antigen (VCA) appear early in the course of the infection, but at the time of clinical symptoms, IgG levels to VCA are quite high. Thus, paired sera at the time of clinical infection do not help diagnose the disease. In contrast, antibod-ies to EB nuclear antigen appear months after the infection. Antibodies to early anti-gen appear in about 70 percent of primary infections and are usually indicative of an acute infection.
The clinical signs of fever, such as enlarged cervical lymph nodes, and red-dened pharynx with whitish exudates (also seen with other viruses) are often mistaken for group A streptococcal pharyngitis. Thus, culture and antibody tests are used to distinguish between the two.
EBV has one unique feature compared with other viruses in that it produces. disease by infecting and transforming B lymphocytes via the CD21 molecule on the surface of B cells. These infected cells multiply like tumor cells, and up to half of the lymphoid cells from the tonsils of EBV-infected patients may be transformed.
The infection is stopped by a T-cell response that kills virus-infected cells and a humoral response that neutralizes free virions. The characteristic “atypical lym-phocytes” seen in this disease are CD8+ cytotoxic T cells that kill EBV-infected B cells.
Most individuals handle EBV infection quite well. However, rare individuals have a specific defect that fails to handle EBV infections and usually die of the disease. More important is the failure to contain EBV infections secondary to immunosup-pressive therapy. For example, the x-linkedlymphoproliferative syndrome that affects males from age 6 months to 20 years is in this category. These patients die of lymphomas, aplastic anemia, or immunodeficiency.
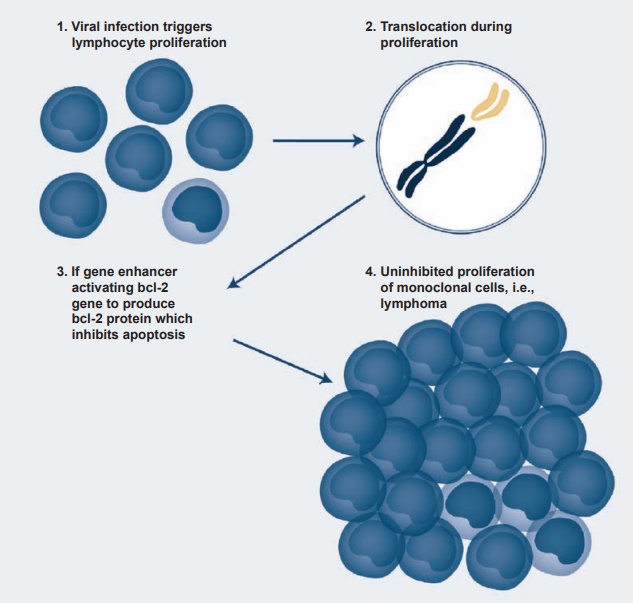
Immunosuppressive therapy may result in EBV reactivation, and about 1 to 10 percent of certain transplants may be com-plicated by EBV-induced lymphoprolifera-tive disease. Similar results are also seen in patients infected with the HIV virus. One of the most interesting effects of EBV infec-tion and transformation has been the occur-rence of Burkitt’s lymphoma. It is endemic in certain African countries (especially in regions of mosquito-borne disease), where it represents 90 percent of childhood can-cers in contrast to 3 percent in developed countries. The link between EBV and Burkitt’s lymphoma was established when it was found that EBV antigens and genome were present in the tumor cells. Since the incidence of Burkitt’s lymphoma follows the same belt as the chronic malaria belt of the region, it has been suggested that the chronic malaria of these children induces EBV-infected lymphoproliferation, leading to chromosomal translocation in the long arms of chromosomes 8 and 14 and even-tually to the activation and translocation of the c-myc oncogene to the active region of the cellular genome (see Figure 4.4).
Viruses have evolved many mecha-nisms to evade the immune response, but one of the key elements in these evasive mechanisms has been their ability to induce a latent status within the cells of the host. Thus, all herpes viruses can remain latent, and no viral antigen is expressed. When there is a change in the host-virus equilib-rium secondary to certain other infections, metabolic disturbances, immunosuppres-sion, or aging, only then does the virus reactivate and cause disease. Examples are herpes simplex in the trigeminal gan-glia, which causes recurrent “cold sores,” or herpes zoster in a dorsal root ganglion, which causes “shingles.”
A second example is antigenic variation or drift in certain viruses. The best example is influenza A, an RNA virus surrounded by a lipid envelope into which two impor-tant proteins (hemagglutinin and neuro-minidase) are inserted. The virus evades the neutralizing antibodies to this virus by constantly modifying the structure of these two proteins by either antigenic drift or antigenic shift. In the former, the change is minor and accounts for minor epidemics of flu during each winter. Antigenic shift is a major change in the structure of those pro-teins, which can cause a major pandemic of devastating proportions. Viewed in this light, the influenza vaccine currently in use is generally protective against common flu epidemics in the world but would not be protective against a major antigenic shift.
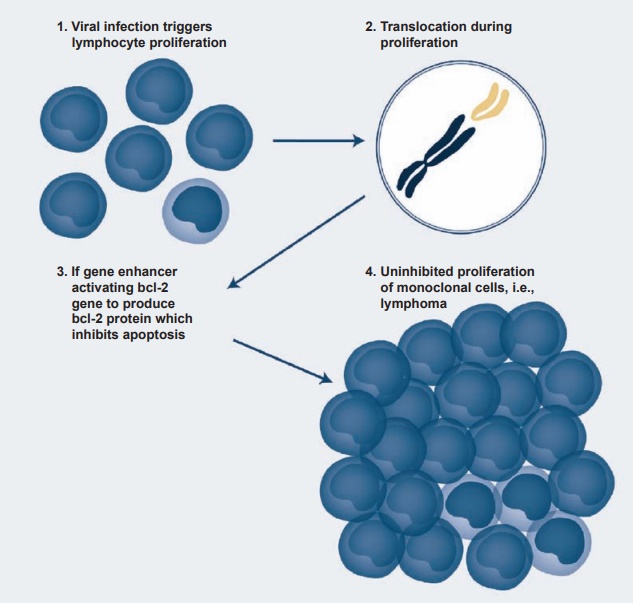
Figure 4.4 A schematic presentation of the steps involved in the transformation of Epstein-Barr virus– infected B cells into a B-cell lymphoma.
A third mechanism is viral persistence in which the virus is not cleared and a chronic infection with persisting virus occurs. For example, HIV persists because it eventually destroys the CD4 T cells needed to kill the virus. Hepatitis C also may persist for years with continuous carriage of the virus in the liver.
While immunological reactions to the virus are generally beneficial to the host, they may also initiate or aggravate tis-sue damage in the host inadvertently. For example, EBV is a polyclonal B-cell activator, and the virus may combine with host antigens to form new antigens. Antibodies to these antigens will not only recognize viral antigens, but some will bind to host antigens as well, thereby possibly initiat-ing an autoimmune response and disease. An example of this type of reaction is the appearance of chronic autoimmune liver disease following chronic hepatitis B infec-tion or immune complex disease such as vasculitis or glomerulonephritis.
Another example of viral bystander damage is dengue fever in which immune enhancement plays an important part. Here, the dengue virus can infect macrophages via Fc receptors, and its capacity to enter the target cell is enhanced if it is bound to IgG antibodies. Thus, the first infection with a specific dengue virus type elicits antibodies to that specific virus. How-ever, cross immunity to the other dengue serotypes is fleeting and lasts only a few weeks. When the individual is exposed to a second dengue virus serotype, the pre-existing antibody to the first type binds to it but does not neutralize the virus. The antibody-enhanced second virus enters the macrophage, setting off a veritable explo-sion of proteases, lymphokines comple-ment activation, coagulation cascade, and vascular permeability factors. The result is a much more severe disease called dengue hemorrhagic fever, whose mortality rate in areas with poor medical facilities and treat-ment may be as high as 50 percent.
Related Topics