Chapter: Modern Pharmacology with Clinical Applications: Drugs Used in Mood Disorders
Treatment of Major Depression: Tricyclic Antidepressants
Tricyclic Antidepressants
In the late 1950s, imipramine
was noted to be effective for the symptomatic treatment of depression. A number
of chemical congeners of imipramine have been synthe-sized and tested for
antidepressant properties; they are collectively known as TCAs. The TCAs are no
longer considered first-line agents in the treatment of depres-sion because of
their prominent side effects and the need to monitor drug blood levels to avoid
toxicity.
Seven TCA drugs are available
in the United States for treatment of major depression. They are generally
categorized as tertiary or secondary amines. Tertiary amines include imipramine
(Tofranil), amitriptyline (Elavil), trimipramine (Surmontil), and doxepin (Sin-equan). Desipramine (Norpramin), nortriptyline (Pam-elor), and protriptyline (Vivactil) are secondary amines.
Clomipramine (Anafranil) also a member of the
tri-cyclic family, possesses similar pharmacology and anti-depressant efficacy.
This agent, however, has Food and Drug (FDA) approval only for use in the
treatment of obsessive-compulsive disorder and is not included in this discussion
of antidepressant drugs.
Maprotiline (Ludiomil) and amoxapine (Asendin) are heterocyclic antidepressant
agents that are not members of the tricyclic family. However, their
pharma-cology is so similar to that of the tricyclic amines that they are
included for discussion purposes with this class of agents. Desipramine and
nortriptyline are major metabolites of imipramine and amitriptyline,
respec-tively.
Mechanism of Action
The precise molecular
mechanism responsible for the antidepressant action of the TCA drugs is
unknown, al-though a number of hypotheses have been generated. Many of these
involve alterations in neurotransmission of norepinephrine or serotonin or
both.
β-Adrenoceptor down-regulation at central nora-drenergic synapses is one popular theory used to ex-plain the antidepressant properties of TCA drugs and other antidepressants. This theory focuses on a cascade of adaptive changes at the noradrenergic synapse that appears to be triggered by inhibition of norepinephrine neuronal reuptake by TCA drugs (Fig. 33.2). Subsensi-tivity in the β-adrenoceptor–coupled adenylyl cyclase system and associated reductions in β-adrenoceptor density appear to be common features of the antide-pressants and of electroconvulsant treatment.

More-over, the time-dependent changes in β-adrenoceptor function parallel the time
delay associated with clinical efficacy of these drugs (2–3 weeks). These
latter findings have added to the attractiveness of this theory. However, at
noradrenergic synapses with multiple adrenoceptors (i.e., α1-, α2-, and β-adrenoceptors), synaptic
transmis-sion through α1-adrenoceptors will likely be enhanced at the same time that
synaptic transmission through α2-and β-adrenoceptors is reduced (Figure 33.3).
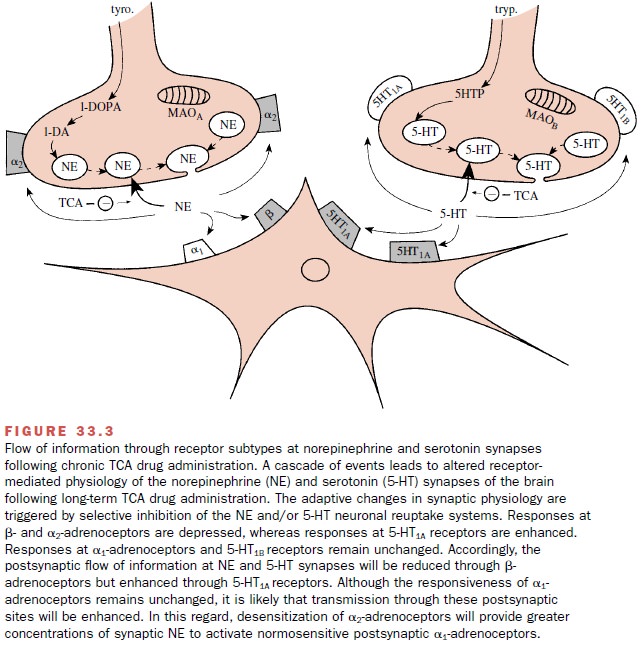
While much emphasis has been
placed on alter-ations in noradrenergic neurotransmission, TCA drugs are not
without effect on serotonin (5-HT) neurotrans-mission. Long-term studies with
TCA drugs in animals have demonstrated postsynaptic supersensitivity to
serotonin (5-HT1A) receptor agonists at serotonin synapses, with an
associated enhancement of serotoner-gic neurotransmission. The sensitization to
5-HT1A ago-nists is mediated in part by an increase in the density
of postsynaptic 5-HT1A receptors. Enhancement of trans- mission
through 5-HT1A receptors appears to be a com-mon phenomenon after
chronic administration of all clinically effective antidepressants and
electroconvul-sive treatment. The occurrence of this 5-HT1A
supersen-sitivity parallels the delayed onset of the therapeutic ac-tions of
these agents (2–3 weeks). These observations lend strong support to the
hypothesis that enhanced serotonergic neurotransmission is required for the
ther-apeutic benefit from TCA drugs.
It is likely that TCA drugs produce their therapeutic benefits by acting at both serotonin and norepinephrine synapses. The literature also supports the notion of an interdependence of these two monoamine systems in the treatment of depression. The time-dependent changes in the flow of synaptic information through in-dividual receptor subtypes within the norepinephrine and serotonin synapses following chronic TCA adminis-tration are summarized in Figure 33.3.
Pharmacokinetics
The TCA drugs are well
absorbed from the gastroin-testinal tract, are extremely lipid soluble, and
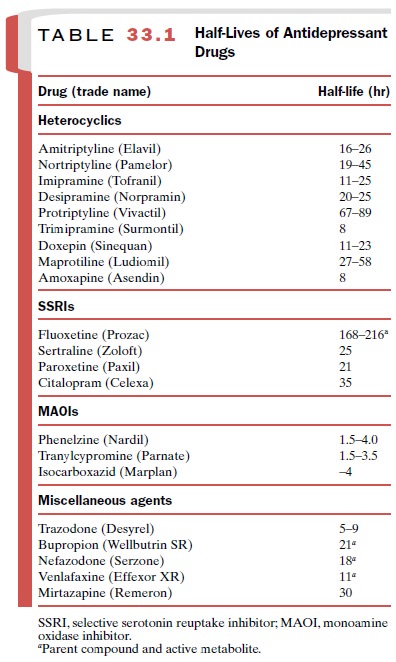
bind ex-tensively to plasma proteins. Their half-lives range from 8 to 89 hours
(Table 33.1). Several days to weeks are re-quired both to achieve steady-state
serum levels and for complete elimination of these agents from the body. Long
half-lives make most of these agents amenable to dosing once a day, generally
at bedtime.
Drug inactivation generally
occurs through oxida-tive metabolism by hepatic microsomal enzymes. Tertiary
amines are converted to secondary amines, which generally possess biological
activity and are fre-quently in serum at levels equal to or greater than that
of the parent tertiary amine. A second route of inacti-vation includes
conjugation of hydroxylated metabo-lites with glucuronic acid.
Adverse Effects
The TCA drugs have lost their
place as first-line therapy for depression because of their bothersome side
effects (Table 33.2) at therapeutic doses and lethal effects in toxic doses. In
addition to their presynaptic effects on the neuronal uptake of norepinephrine
and serotonin, they block several postsynaptic receptors. They are po-tent
cholinergic muscarinic receptor antagonists, result-ing in symptoms such as dry
mouth, constipation, tachy-cardia, blurred vision and urinary retention.
Blockade of histamine receptors (H1) often results in sedation and
weight gain. Antagonism of α1-adrenoceptors in the vas-culature can cause orthostatic
hypotension.

TCA drugs have potent
membrane-stabilizing prop-erties similar to those of quinidine. Conduction is
slowed throughout the heart, and serious ventricular ar-rhythmias may develop
in patients with preexisting con-duction abnormalities at therapeutic doses and
in all pa-tients at toxic doses. At therapeutic doses, the TCA drugs lower the
seizure threshold and at toxic doses can cause life-threatening seizures.
Maprotiline has a greater potential for reducing the seizure threshold and
should not be used in patients with a seizure disorder. Amoxapine has dopamine
receptor antagonist proper-ties and can
induce extrapyramidal side effects, gynecomastia, lactation, and neuroleptic
malignant syndrome. To increase tolerance to the anti-cholinergic and
orthostatic effects of the TCA drugs, the dose is usually titrated with
increasing increments over the first few weeks of therapy.
Drug Interactions
Multiple drug interactions
can occur with the TCA drugs. Because of their high degree of binding to plasma
proteins, competition for binding sites can exist be-tween TCAs and phenytoin,
aspirin, phenothiazines, and other agents that also display avid plasma
protein-binding characteristics. Elevation in the serum level of TCAs (with
corresponding toxicity) can occur following the administration of one of these
second drugs. Elevations in the serum TCA level also can occur fol-lowing
inhibition of hepatic TCA metabolism by an-tipsychotics, methylphenidate, oral
contraceptives, and some SSRIs.
Tricyclic antidepressant
drugs can prevent the action of antihypertensive drugs, such as guanethidine
and clonidine. This antagonistic action is related to the pri-mary (inhibition
of neuronal reuptake) and secondary (adaptive changes) effects of TCA drugs at
noradrener-gic synapses. A more serious but rare interaction exists between TCA
drugs and MAOIs. While both classes of drugs are effective in the treatment of
major depres-sion, simultaneous administration of a drug from each class can
result in severe CNS toxicity (hyperpyrexia, convulsions, and coma). In
TCA-resistant patients, it is advisable to discontinue the TCA drug for 2 to 3
weeks before initiation of a MAOI agent. Finally, TCA drugs potentiate the
sedative effects of alcohol, and patients must be cautioned about this
interaction.
Therapeutic Drug Monitoring
Safe and effective use of the
TCA drugs requires moni-toring of serum levels. The importance of this
monitor-ing is based on the relatively narrow range between therapeutic and
toxic doses (therapeutic index of 3) of each agent. While annoying side effects
(sedation, dry mouth, constipation) begin to occur at subtherapeutic serum
concentrations, life-threatening cardiac and CNS effects develop in a dose
dependent fashion above serum levels of 500 ng/mL. The metabolism and elimi-nation
rates vary 10- to 30-fold among individuals tak-ing TCA drugs. For this reason,
it is estimated that only 50% of the patients receiving a standard dose of a
TCA drug would achieve an optimal therapeutic serum con-centration. Of
additional concern, 3 to 5% of patients will be deficient in hepatic enzymes
that metabolize the TCA drugs and may develop life-threatening serum lev-els on
standard doses. Therefore, steady-state serum lev-els of TCA drugs (drawn 10 to
12 hours after the last dose) are monitored to avoid toxicity, monitor
compli-ance, and optimize the therapeutic response.
Related Topics