Chapter: Modern Pharmacology with Clinical Applications: Drugs Used in Mood Disorders
Treatment of Major Depression: Miscellaneous Antidepressants
Miscellaneous Antidepressants
Venlafaxine
Venlafaxine (Effexor) inhibits the reuptake of both
serotonin and norepinephrine at their respective presy-naptic sites. This drug
does not have significant effects at muscarinic, histamine, or α-adrenergic receptors and
therefore is devoid of many of the side effects associ-ated with the TCAs.
Venlafaxine and its active metabo- lite, O-desmethyl-venlafaxine,
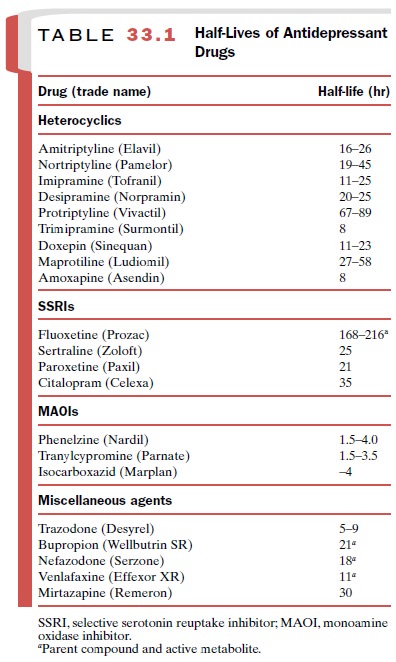
have half lives of 5 and 11 hours respectively, so dosing twice a day is
necessary. However, an extended release preparation (Effexor XR) now allows
for once-daily dosing and better toler-ance. Venlafaxine has a side effect
profile similar to that of the SSRIs (Table 33.2). Higher doses of venlafaxine
result in modest increases in blood pressure in approxi-mately 5% of patients.
Venlafaxine has minimal effects on the cytochrome P450 enzyme system.

Bupropion
Bupropion (Wellbutrin) is a pharmacologically
unique antidepressant, since it is a weak inhibitor of both do-pamine and
norepinephrine neuronal reuptake. How-ever, its actual antidepressant activity
is not well under-stood. Bupropion is generally well tolerated and does not
block muscarinic, histaminergic, or adrenergic re-ceptors. Unlike the SSRIs and
venlafaxine, bupropion does not cause sexual side effects. However, it can
cause CNS stimulation, including restlessness and insomnia. High doses of
bupropion, given as its original formula-tion, were associated with a risk of
seizures in 0.4% of patients. However, this risk is lower with slow-release
bupropion (Wellbutrin SR). This
formulation still re-quires dosing twice a day, and bupropion is
contraindi-cated in patients with a history of seizures. Bupropion inhibits the
cytochrome P450 2D6 isoenzyme and may elevate blood levels of drugs metabolized
by this route.
Mirtazapine
Mirtazapine (Remeron) enhances both serotonergic and noradrenergic neurotransmission. By blocking presynaptic α2-adrenoceptors, mirtazapine causes re-lease of norepinephrine. Indirectly, through noradrener-gic modulation of serotonin systems, mirtazapine also causes increased release of serotonin.
It is an antagonist at the 5-HT2A, 5HT2C,
5-HT3, and histamine receptors but has minimal affinity for muscarinic
or α1-receptors. Mirtazapine does
not inhibit neuronal reuptake of sero-tonin or norepinephrine. Weight gain and
sedation are common side effects (Table 33.2); sedation necessitates dosing at
bedtime. Mirtazapine does not have signifi-cant effects on cytochrome P450
isoenzymes.
Trazodone
Trazodone (Desyrel) was introduced in the early
1980s as a second-generation antidepressant. It blocks the neuronal reuptake of
serotonin and is an antagonist at the 5HT2-receptor. Also, its major
metabolite, m-chlorophenylpiperazine
(mCPP), is a postsynaptic sero-tonin receptor agonist. When compared to the
TCAs, trazodone is relatively free of antimuscarinic side ef-fects, but it does
block the α-adrenoceptor. Common side effects include marked sedation,
dizziness, ortho-static hypotension, and nausea (Table 33.2). Priapism is an
uncommon but serious side effect requiring surgical intervention in one-third
of the cases reported. Because of trazodone’s sedating quality, it is often
used in low doses to counter the insomnia associated with the newer
antidepressants, such as the SSRIs.
Nefazodone
Although nefazodone (Serzone) is structurally related to
trazodone, it is less sedating. It does not block α1-adrenoreceptors, and its use
is not associated with pri-apism. Nefazodone inhibits the neuronal reuptake of
serotonin and blocks 5HT2A receptors. Its short half-life requires
dosing twice a day (Table 33.1). Nefazodone is not associated with weight gain
or sexual dysfunction. It inhibits the cytochrome P450 3A4 isoenzyme that is
re-sponsible for 50% of known oxidative metabolism, and therefore, nefazodone
can elevate levels of drugs de-pendent on this pathway for metabolism.
Related Topics