Chapter: Modern Pharmacology with Clinical Applications: Insulin and Oral Drugs for Diabetes Mellitus
Sulfonylureas - Oral Agents For Treating Diabetes Mellitus
Sulfonylureas
Sulfonylureas are the most
widely prescribed drugs in the treatment of type II diabetes mellitus. The
initial sulfonylureas were introduced nearly 50 years ago and were derivatives
of the antibacterial sulfonamides. Although their structural similarities to
the sulfon-amide antibacterial agents are readily apparent, the sul-fonylureas
possess no antibacterial activity.
Mechanism of Action
The primary mechanism of
action of the sulfonylureas is direct
stimulation of insulin release from the pancreatic β-cells. In the presence
of viable pancreatic β-cells, sul-fonylureas enhance the release of endogenous
insulin, thereby reducing blood glucose levels. At higher doses, these drugs
also decrease hepatic glucose production, and the second-generation
sulfonylureas may possess additional extrapancreatic effects that increase insulin
sensitivity, though the clinical significance of these phar-macological effects
is unclear. These mechanisms are summarized in Table 67.3.
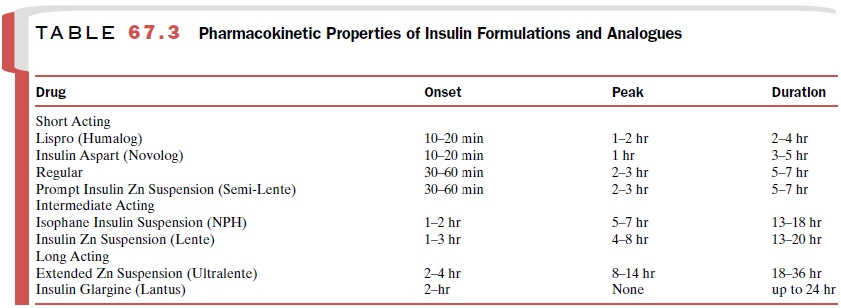
The sulfonylureas are ineffective for the manage-ment of type
I and severe type II diabetes mellitus, since the number of viable β-cells in
these forms of diabetes is small. Severely obese diabetics often respond poorly
to the sulfonylureas, possibly because of the insulin re-sistance that often
accompanies obesity.
The Sulfonylurea Receptor
The sulfonylurea receptor was
identified as an adeno-sine triphosphate (ATP) sensitive potassium (KATP)
channel that is present on the β-cell membrane surface. Closure of these KATP
channels causes β-cell membrane depolarization and triggers the opening of
voltage-dependent calcium channels. The influx of calcium into the β-cell
triggers insulin granule fusion to the β-cell membrane and insulin release. The
intracellular levels of ATP and adenosine diphosphate (ADP) modulate the
activity of the KATP channel, depending on the avail-ability of
glucose.
The activity of the KATP
channels is modulated by the direct binding of sulfonylureas to a specific
subunit of the KATP channel called SUR1. SUR1 is a member of the K+
inwardly rectifying (Kir) 6.0 subfamily of pro-teins and can bind
nucleotides and sulfonylureas with high affinity. Four SUR1 subunits form a
complex with four subunits from the Kir 6.2 subfamily and create the pore for K+
permeation in the pancreatic β-cell. Sulfonylurea binding to SUR1
directly promotes the closure of these KATP channels, lowering the
threshold for glucose-dependent insulin release. Diazoxide also binds to SUR1
but keeps the KATP channels open, raising the threshold for glucose-stimulated
insulin secretion and sometimes causing hyperglycemia in patients.
Absorption, Metabolism, and Excretion
Sulfonylureas are readily
absorbed from the gastroin-testinal tract following oral administration but
undergo varying degrees and rates of metabolism in the liver and/or kidney;
some metabolites possess intrinsic hy-poglycemic activity. Thus, the biological
half-lives of the sulfonylureas vary greatly, and a comparison of the drug
half-life with the observed duration of action does not always show a good
correlation. Sulfonylureas and their metabolites are excreted either renally or
in the feces.
Clinical Uses
Sulfonylureas are generally
effective in individuals with mild to moderate type II diabetes. The chance for
suc-cessful glycemic control with sulfonylureas is poor in di-abetic patients
requiring more than 40 units of insulin per day. When beginning therapy with
one of these drugs, a low to intermediate dose is given initially and then
gradually increased until the dosage results in nor-moglycemia. Once the
maximum recommended dosage for a particular sulfonylurea is reached, further
increas-ing the dose will not improve glycemic control.
Adverse Effects and Drug Interactions
The most common adverse
effect associated with sul-fonylurea administration is hypoglycemia, which may
be provoked by inadequate calorie intake (e.g., skipping a meal), or increased
caloric needs (e.g., increased phys-ical activity). Collectively, sulfonylureas
also tend to cause weight gain, which is undesirable in individuals
who already are obese. Some
of this weight can be due to fluid retention and edema. Less common adverse
re-actions include muscular weakness, ataxia, dizziness, mental confusion, skin
rash, photosensitivity, blood dyscrasias, and cholestatic jaundice.
Occasionally, per-sons who display drug sensitivities to sulfa-containing
antibiotics show a cross-reactivity to the sulfonylureas. In this situation, a
nonsulfonylurea insulin secretagogue can be used (if desired), such as
repaglinide or nateglin-ide (discussed later). Sulfonylureas are not used in
ges-tational diabetes, which is generally managed by a com-bination of
intensive diet control and insulin.
Since diabetic patients with
renal or hepatic disease are particularly vulnerable to hypoglycemia, the
sul-fonylurea compounds should be avoided in these indi-viduals. A decrease in
alcohol tolerance also has been observed in some patients taking sulfonylurea
com-pounds. Since sulfonylureas are highly bound to plasma proteins and are extensively
metabolized by microso-mal enzymes, coadministration of drugs capable of
dis-placing them from their protein binding sites or inhibit-ing their
metabolism (e.g., sulfonamide antibacterials, propranolol, salicylates,
phenylbutazone, chlorampheni-col, probenecid, and alcohol) also may potentiate
hypo-glycemia.
First-Generation Sulfonylureas
The first-generation
sulfonylureas are not frequently used in the modern management of diabetes
mellitus because of their relatively low specificity of action, de-lay in time
of onset, occasional long duration of action, and a variety of side effects.
They also tend to have more adverse drug interactions than the
second-gener-ation sulfonylureas. They are occasionally used in pa-tients who
have achieved previous adequate control with these agents.
Acetohexamide (Dymelor) is the only sulfonylurea with
uricosuric activity, an action that may be of benefit in diabetic patients who
also have gout.
Chlorpropamide (Diabinese) has a relatively slow onset
of action, with its maximal hypoglycemic poten-tial often not reached for 1 or
2 weeks. Similarly, several weeks may be required to eliminate the drug after
dis-continuation of therapy. This drug can cause flushing, particularly when
taken with alcohol, and can also cause hyponatremia. This effect has been
employed to treat some patients who have partial central diabetes in-sipidus,
an unrelated condition due to a pituitary ADH deficiency.
Tolazamide (Tolinase) is an orally effective
hypo-glycemic drug that causes less water retention than do the other compounds
in this class.
Tolbutamide (Orinase) is a relatively short-acting
compound that may be useful in patients who are prone to hypoglycemia.
Second-Generation
Sulfonylureas
The second-generation
sulfonylureas display a higher specificity and affinity for the sulfonylurea
receptor and more predictable pharmacokinetics in terms of time of onset and
duration of action, and they have fewer side effects. Second-generation
sulfonylureas may also exert mild diuretic effects on the kidney and are highly
protein bound, primarily through nonionic bind-ing (in contrast to the ionic
binding observed with the first-generation compounds).
Glyburide (DiaBeta, Micronase, Glynase), also known
as glibenclamide, is approximately 150 times as potent as tolbutamide on a
molar basis and twice as potent as glipizide (discussed later). Glyburide is
completely me-tabolized in the liver to two weakly active metabolites before
excretion in the urine. Its average duration of ac-tion is 24 hours.
Glipizide (Glucotrol) is similar to glyburide, but
it is metabolized by the liver to two inactive metabolites; these metabolites
and glipizide are renally excreted.
Glimepiride (Amaryl) is metabolized to at least one
active metabolite. It is quickly absorbed from the gas-trointestinal tract
within an hour of oral administration and excreted in the urine and feces. Its
half-life varies from 5 to 9 hours depending on the frequency of multi-ple
dosing.
Related Topics